Un ratón transgénico que expresa un transgén que comprende una secuencia de ADN que codifica una Proteína Precursora Amiloide (APP) heteróloga que comprende la mutación Arctic (E693G) y una mutación patógena adicional de la EA (enfermedad de Alzheimer) seleccionada entre el grupo que consiste en KM670/671DF,
KM670/671DY, KM670/671EF, KM670/671EY, KM670/671NL, KM670/671NY, KM670/671NF, KM670/671KL, KM670/671DL and KM670/671EL, y sin mutaciones de APP adicionales, en el que dicho transgén se une de manera operable a un promotor eficaz para la expresión de dicho gen en el tejido cerebral de dicho ratón, lo que da como resultado cantidades crecientes de agregados A13 solubles intracelulares, que incluyen péptidos Aß
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/SE2005/000383.
A01K67/027NECESIDADES CORRIENTES DE LA VIDA. › A01AGRICULTURA; SILVICULTURA; CRIA; CAZA; CAPTURA; PESCA. › A01KCRÍA DE ANIMALES; AVICULTURA; APICULTURA; PISCICULTURA; PESCA; ANIMALES PARA CRIA O REPRODUCCIÓN, NO PREVISTOS EN OTRO LUGAR; NUEVAS VARIEDADES DE ANIMALES. › A01K 67/00 Cría u obtención de animales, no prevista en otro lugar; Nuevas razas de animales. › Nuevas razas de vertebrados.
Clasificación antigua:
A01K67/027A01K 67/00 […] › Nuevas razas de vertebrados.
Países PCT: Austria, Bélgica, Suiza, Alemania, Dinamarca, España, Francia, Reino Unido, Grecia, Italia, Liechtensein, Luxemburgo, Países Bajos, Suecia, Mónaco, Portugal, Irlanda, Eslovenia, Finlandia, Rumania, Chipre, Lituania.
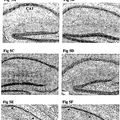
La presente invención se refiere a un modelo de ratón transgénico de la enfermedad de Alzheimer y a los trastornos neurológicos relacionados. La presente invención se refiere también a un procedimiento para producir dicho ratón transgénico, y a los procedimientos de cribado de agentes terapéuticos o diagnósticos útiles en el tratamiento o el diagnóstico de la enfermedad de Alzheimer. Antecedentes de la invención ES 2 367 310 T3 La enfermedad de Alzheimer (EA) es un trastorno degenerativo progresivo e irreversible que produce discapacidades cognitivas, de memoria y de comportamiento. Es la causa más habitual de demencia entre los ancianos afectando aproximadamente al 5% de la población por encima de los 65 años y al 20% por encima de los 80 años de edad La EA se caracteriza por un comienzo insidioso y un deterioro progresivo en múltiples funciones cognitivas. La neuropatología implica depósitos proteínicos argirofílicos extracelulares e intracelulares. Los depósitos extracelulares, denominados placas neuríticas, están constituidos principalmente por péptidos de beta amiloide (Aß) rodeados por neuritas distróficas (procesos neuronales hinchados, distorsionados). Los péptidos Aß contenidos en estos depósitos extracelulares son fibrilares en su carácter con una estructura en lámina ß plegada. Se puede teñir el Aß en estos depósitos con algunos colorantes, por ejemplo, Rojo Congo y muestra una ultraestructura fibrilar: Estas características, adoptadas por los péptidos Aß en su estructura fibrilar de placas neuríticas son la base del término genérico amiloide. Frecuentes placas neuríticas y depósitos de ovillos neurofibrilares en el cerebro son criterios diagnósticos de la EA, que se realizan cuando el paciente ha muerto. Los cerebros EA muestran también atrofia macroscópica del cerebro, pérdida de células nerviosas, inflamación local (microgliosis y astrocitosis) y a menudo, angiopatía amiloide congofílica (AAC) en las paredes de los vasos cerebrales. Dos formas de péptidos Aß, Aß40 y Aß42, son las especies dominantes en las placas neuríticas de EA (Masters y col., 1985), aunque Aß40 es la especie destacada en el amiloide cerebrovascular asociada con la EA (Glenner y Wong, 1984). Las actividades enzimáticas permiten que se formen continuamente estas Aß a partir de una proteína más grande denominada la proteína precursora amiloide (AAP) en sujetos sanos y que padecen EA en todas las células del cuerpo. Dos episodios principales de procesamiento de AAP (las actividades ß y -secretasa, permiten la producción de péptido Aß mediante escisión enzimática, mientras que una tercera actividad, denominada secretasa, evita el péptido Aß mediante la escisión en el interior de la secuencia del péptido Aß (revisado en Selkoe, 1994; documento US5604102). El Aß42 es un péptido que tiene una longitud de cuarenta y dos aminoácidos, es decir, dos aminoácidos más largo en el extremo C, en comparación con Aß40 E péptido Aß42 es más hidrófobo, y se agrega más fácilmente en estructuras más grandes de péptidos Aß, tales como dímeros Aß, tetrámeros Aß, oligómeros Aß, protofibrillas Aß o fibrillas Aß son hidrófobas e insolubles, mientras que las otras estructuras son todas menos hidrófobas y solubles. Todas estas estructuras moleculares mayores de péptidos Aß se definen individualmente basándose en su apariencia biofísica y estructural, por ejemplo, en el microscopio electrónico, y sus características bioquímicas, por ejemplo mediante análisis de cromatografía de exclusión por tamaño/transferencia western. Estos péptidos Aß, particularmente Aß42 se ensamblarán gradualmente en diversas estructuras moleculares mayores de Aß durante toda la vida. La EA, que es un trastorno fuertemente dependiente de la edad, se producirá antes en el transcurso de la vida si su proceso de ensamblaje se produce más rápidamente en el cerebro del individuo. Esto es el punto central de la hipótesis de la cascada amiloide de la EA, que requiere el procesamiento de APP, los niveles de Aß42 y su ensamblaje en estructuras moleculares mayores son la causa central de toda la patogénesis de la EA. Todas las diferentes neuropatologías de la EA cerebral y los síntomas de la EA tales como la demencia son, de alguna manera, producidos por los péptidos Aß o sus formas de ensamblaje. La evidencia principal de la hipótesis de la cascada amiloide procede de los estudios genéticos de individuos de familias que padecen un comienzo temprano de la EA familiar como rasgo dominante. Estos estudios han desvelado que mutaciones raras en el gen de la AAP son suficientes para generar formas graves de EA. Las mutaciones se agrupan en y alrededor de Val 717 ligeramente en la dirección 3 del extremo C de Aß1-42 (Goate y col., 1991, Chartier-Harlan, y col., 1991, Murrell, y col., 1991) y una única mutación doble (670-671) inmediatamente en la dirección 5 del extremo N de Aß en una familia Swedish (Mullan, y col., 1992; US5795963). Las mutaciones de AAP, que enmarcan la secuencia del péptido Aß (la mutación Swedish; Citron, y col., 1992, Cai y col., 1993), o que aumentan la relación de producción de Aß42/Aß40 y que generan también péptidos Aß que se extienden en el extremo C para incorporar la mutación patogénica en el péptido Aß, por ejemplo, Aß50 (las mutaciones 717 están en la posición 46; Suzuki y col., 1994; Roher y co., 2003). De esta manera, las mutaciones 717, además de Aß40 natural y Aß42 natural, generan también los péptidos London Aß (V717I) y los péptidos Aß Indiana (V717F, Aß46 y formas más largas de Aß) que forman rápidamente fibrilla Aß. En contraste, la mutación Sueca genera únicamente 2 ES 2 367 310 T3 niveles crecientes de péptidos Aß40 y Aß42 naturales. El comienzo temprano de la EA familiar se produce más a menudo por mutaciones en presenilina 1 (en el cromosoma 14; documentos US5986054; US5840540; US5449604) y en algunas casos por mutaciones en presenilina 2 (cromosoma 1). La presenilina 1 y la presenilina 2 son proteínas transmembrana politópicas que, junto las otras tres proteínas nicastrina, aph1 y pen-2, constituyen el núcleo funcional necesario del complejo de la -secretasa que permite la formación del péptido Aß mediante la escisión enzimática de AAP (Edbauer y col., 2003). Todas las mutaciones patógenas de la EA en proteínas presenilina 1 y presenilina 2 aumentan significativamente la producción en exceso de Aß1-42 (Schuener y col., 1996). La apolipoproteína E (ApoE) es, además de la edad, el factor de riesgo más importante para el comienzo tardío de la EA. Existen tres variantes de la proteína ApoE en seres humanos, debido a una única sustitución de aminoácidos en la proteína ApoE. La variante ApoE4 confiere un creciente riesgo de EA, mientras que la variante ApoE2 es protectora en comparación con la variante ApoE3 predominante (Strittmatter y col., 1993; Corder y col., 1993). Estos cambios de proteínas no son determinísticos, pero confieren una susceptibilidad creciente o decreciente a desarrollar la EA en una población. La capacidad de las variantes de ApoE de facilitar la deposición amiloide en modelos APP de ratones transgénicos de EA es mayor para ApoE4, intermedia para ApoE3 y menor para ApoE2, sugiriendo que el mecanismo patógeno de la EA es para potenciar el ensamblaje del péptido Aß y/o la deposición amiloide (Fagan y col., 2002). Otras proteínas tales como a1-antiquimotripsina (Nilsson y col., 2001) y ApoJ/clusterina (DeMattos y col., 2002), potencian también el ensamblaje del péptido Aß y/o la deposición amiloide en ratones APP transgénicos, de manera simular a ApoE. Neprilisina (NEP) y la enzima degradadora de la insulina (IDE) degradan los péptidos Aß y están implicadas probablemente en la EA. Sin embargo, ningún genetista humano ha probado que estas proteínas estén implicadas en la EA. Una característica esencial es comprender mejor como el aumento de los niveles de Aß o de sus agregados producen demencia y pérdida funcional en pacientes con EA. Se ha creído desde hace mucho tiempo que las fibrillas amiloides insolubles, el principal componente de la placa neurítica, son las especies patógenas en el cerebro con EA. Se ha demostrado que elevadas concentraciones de fibrillas Aß son citotóxicas en modelos de cultivo celular de células nerviosas en el cerebro (Pike y col., 1991; Lorenzo y Yankner y col., 1994). Sin embargo la hipótesis de la fibrilla amiloide como la principal especie neurotóxica es inconsistente con la mala correlación entre la densidad de la placa neurítica y el marco de la demencia por EA y también con pequeños signos de neurodegeneración en ratones APP transgénicos actuales. Se podrían hacer desaparecer la unión las especies neurotóxicas solubles de intermedios de Aß y sus sitios subcelulares de formación y distribución... [Seguir leyendo]
Reivindicaciones:
1. Un ratón transgénico que expresa un transgén que comprende una secuencia de ADN que codifica una Proteína Precursora Amiloide (APP) heteróloga que comprende la mutación Arctic (E693G) y una mutación patógena adicional de la EA (enfermedad de Alzheimer) seleccionada entre el grupo que consiste en KM670/671DF, KM670/671DY, KM670/671EF, KM670/671EY, KM670/671NL, KM670/671NY, KM670/671NF, KM670/671KL, KM670/671DL and KM670/671EL, y sin mutaciones de APP adicionales, en el que dicho transgén se une de manera operable a un promotor eficaz para la expresión de dicho gen en el tejido cerebral de dicho ratón, lo que da como resultado cantidades crecientes de agregados A13 solubles intracelulares, que incluyen péptidos Aß. 2. El ratón transgénico de acuerdo con la reivindicación 1, en el que el transgén se integra en el ADN genómico. 3. El ratón transgénico de acuerdo con cualquiera de las reivindicaciones 1 ó 2 en el que la APP endógena se expresa o no se expresa. 4. El ratón transgénico de acuerdo con cualquiera de las reivindicaciones 1-3, en el que dicha mutación patógena adicional de la EA es la mutación KM670/671NL de APP. 5. El ratón transgénico de acuerdo con la reivindicación 4, en el que el ratón transgénico expresa únicamente un transgén. 6. El ratón transgénico de acuerdo con cualquiera de las reivindicaciones 1-5, que comprende adicionalmente una construcción directora integrada de manera homóloga para al menos uno de los genes de la neprilisina o que degrada la insulina (IDE), que perturba estos genes mediante la ablación del gen (inactivación) y potencia los niveles de los péptidos Aß-40 y/o Aß-42 Arctic. 7. Un procedimiento de producción del ratón transgénico de acuerdo con cualquiera de las reivindicaciones 1-6, que comprende integrar en el ADN genómico un transgén que comprende una secuencia de ADN que codifica una Proteína Precursora Amiloide (APP) que comprende la mutación Arctic (E693G) y una mutación patógena adicional de la EA (enfermedad de Alzheimer) seleccionada entre el grupo que consiste en KM670/671DF, KM670/671DY, KM670/671EF, KM670/671EY, KM670/671NL, KM670/671NY, KM670/671NF, KM670/671KL, KM670/671DL y KM670/671EL y sin mutaciones de APP adicionales, en el que dicho transgén se une de manera operable a un promotor eficaz para la expresión de dicho gen en el tejido cerebral de dicho ratón. 8. El procedimiento de acuerdo con la reivindicación 7 en el que la APP endógena se prepara no expresiva. 9. El procedimiento de acuerdo con cualquiera de las reivindicaciones 7 u 8, en el que dicha mutación patógena de la EA adicional es la mutación KM670/671NL de APP. 10. El procedimiento de acuerdo con cualquiera de las reivindicaciones 7-9, que comprende adicionalmente integrar de manera homóloga una construcción directora para al menos uno de los genes de la enzima neprilisina o que degrada la insulina (IDE). 11. Un procedimiento de cribado, en el que el ratón transgénico de acuerdo con cualquiera de las reivindicaciones 1-6 se usa para cribar los agentes útiles para tratar, evitar o inhibir la enfermedad de Alzheimer. 12. Un procedimiento de cribado, en el que el ratón transgénico de acuerdo con cualquiera de las reivindicaciones 1-6 se usa para cribar los agentes de diagnóstico de la enfermedad de Alzheimer. 14 ES 2 367 310 T3 ES 2 367 310 T3 16 ES 2 367 310 T3 17 ES 2 367 310 T3 18 ES 2 367 310 T3 19 ES 2 367 310 T3 ES 2 367 310 T3 21 ES 2 367 310 T3 22 ES 2 367 310 T3 23 ES 2 367 310 T3 24
Patentes similares o relacionadas:
PROCEDIMIENTOS Y COMPOSICIONES INMUNOLÓGICAS PARA EL TRATAMIENTO DE LA ENFERMEDAD DE ALZHEIMER, del 23 de Noviembre de 2011, de THE GOVERNING COUNCIL OF THE UNIVERSITY OF TORONTO: Un péptido representado por la fórmula
(A)n -- (Th)m -- (B)o -- Abeta -- (C)p
en la que cada A, B y C son un resto de aminoácido o una secuencia de restos […]
MÉTODO DE DIAGNÓSTICO IN VITRO DE LA ENFERMEDAD DE ALZHEIMER MEDIANTE UN ANTICUERPO MONOCLONAL, del 8 de Noviembre de 2011, de CONSEJO SUPERIOR DE INVESTIGACIONES CIENTIFICAS: Método de diagnóstico in vitro de la enfermedad de Alzheimer medante un anticuerpo monoclonal. Dicho anticuerpo es capaz de unire al menos a los aminoácidos 12-16 del […]
COMPOSICIÓN DE PÉPTIDO INMUNOGÉNICO PARA LA PREVENCIÓN Y EL TRATAMIENTO DE LA ENFERMEDAD DE ALZHEIMER, del 6 de Junio de 2011, de UNITED BIOMEDICAL, INC.: Un inmnunógeno del péptido representado por una de las siguientes fórmulas: (A)n-(fragmento N-terminal del péptido Aß1-42)-(B)o-(Th)m-X; o […]
POLIPÉPTIDOS DERIVADOS DE PROTEÍNA PRECURSORA AMILOIDE (APP) Y SUS USOS, del 31 de Marzo de 2011, de THE OPEN UNIVERSITY: Una composición farmacéutica que comprende: (a) un compuesto con la fórmula: X1 es un grupo acilo representado por la fórmula: X2 es H o un grupo acilo representado […]
COMPOSICION DE PÈPTIDO INMUNOGENO PARA LA PREVENCION Y TRATAMIENTO DE LA ENFERMEDAD DE ALZEHIMER, del 20 de Diciembre de 2010, de UNITED BIOMEDICAL, INC.: Un péptido inmunógeno representado por una de las siguientes fórmulas: en donde cada A es independientemente un aminoácido; (A)n-(fragmento N-terminal […]
ANIMALES TRANSGENICOS QUE PRESENTAN TRANSTORNOS SERIOS ASOCIADOS A LA ENFERMEDAD DE ALZHEIMER, del 26 de Noviembre de 2010, de AVENTIS PHARMA S.A.: Animal no humano que presenta una secuencia de ácidos nucleicos que codifica una proteína presenilina 1 que lleva en el estado homocigoto […]
METODOS PARA LA IDENTIFICACION DE LOS COMPUESTOS INHIBIDORES DE LA LIBERACION DEL PEPTIDO BETA-AMILOIDE (BETAAP), del 21 de Octubre de 2010, de ELAN PHARMACEUTICALS, INC.
THE BRIGHAM AND WOMEN'S HOSPITAL, INC.
ELI LILLY AND COMPANY: Método in vitro para identificar inhibidores de la producción del péptido ß-amiloide (ßAP), comprendiendo dicho método cultivar células de mamífero en un […]
MODELOS DE SELECCIÓN IN VIVO PARA EL TRATAMIENTO DE LA ENFERMEDAD DE ALZHEIMER Y OTROS TRASTORNOS RELACIONADOS CON QPCT, del 17 de Enero de 2012, de PROBIODRUG AG: Animal transgénico no humano que sobreexpresa glutaminil ciclasa que comprende células que contienen un transgén de ADN que codifica para glutaminil ciclasa.
Utilizamos cookies para mejorar nuestros servicios y mostrarle publicidad relevante. Si continua navegando, consideramos que acepta su uso. Puede obtener más información aquí. .
 ANIMALES TRANSGENICOS QUE PRESENTAN TRANSTORNOS SERIOS ASOCIADOS A LA ENFERMEDAD DE ALZHEIMER, del 26 de Noviembre de 2010, de AVENTIS PHARMA S.A.: Animal no humano que presenta una secuencia de ácidos nucleicos que codifica una proteína presenilina 1 que lleva en el estado homocigoto […]
ANIMALES TRANSGENICOS QUE PRESENTAN TRANSTORNOS SERIOS ASOCIADOS A LA ENFERMEDAD DE ALZHEIMER, del 26 de Noviembre de 2010, de AVENTIS PHARMA S.A.: Animal no humano que presenta una secuencia de ácidos nucleicos que codifica una proteína presenilina 1 que lleva en el estado homocigoto […]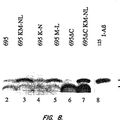 METODOS PARA LA IDENTIFICACION DE LOS COMPUESTOS INHIBIDORES DE LA LIBERACION DEL PEPTIDO BETA-AMILOIDE (BETAAP), del 21 de Octubre de 2010, de ELAN PHARMACEUTICALS, INC.
THE BRIGHAM AND WOMEN'S HOSPITAL, INC.
ELI LILLY AND COMPANY: Método in vitro para identificar inhibidores de la producción del péptido ß-amiloide (ßAP), comprendiendo dicho método cultivar células de mamífero en un […]
METODOS PARA LA IDENTIFICACION DE LOS COMPUESTOS INHIBIDORES DE LA LIBERACION DEL PEPTIDO BETA-AMILOIDE (BETAAP), del 21 de Octubre de 2010, de ELAN PHARMACEUTICALS, INC.
THE BRIGHAM AND WOMEN'S HOSPITAL, INC.
ELI LILLY AND COMPANY: Método in vitro para identificar inhibidores de la producción del péptido ß-amiloide (ßAP), comprendiendo dicho método cultivar células de mamífero en un […]