Procedimiento para preparar un producto intermedio sintético para la preparación de nucleósidos ramificados.
Un procedimiento para preparar un compuesto de Fórmula II **Fórmula**
en la que R1 es alquilo de cadena lineal o de cadena ramificada,
cicloalquilo, alquenilo, alquinilo, alenilo, CF3, ciano,arilo, aralquilo, o heterocíclico, que comprende
a) proporcionar un compuesto de estructura (i) o (ii) **Fórmula**
en donde OR es un grupo lábil y X es halógeno;
b) poner en contacto el compuesto con un agente de fluoración bajo condiciones anhidras; y
c) eliminar el grupo protector de hidroxilo del producto de la etapa b).
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/US2006/048769.
Solicitante: IDENIX PHARMACEUTICALS, INC..
Nacionalidad solicitante: Estados Unidos de América.
Dirección: 320 Bent Street, Floor 4 Cambridge, MA 02141 ESTADOS UNIDOS DE AMERICA.
Inventor/es: MAYES,BENJAMIN ALEXANDER, MOUSSA,ADEL.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C07H15/24 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07H AZUCARES; SUS DERIVADOS; NUCLEOSIDOS; NUCLEOTIDOS; ACIDOS NUCLEICOS (derivados de ácidos aldónicos o sacáricos C07C, C07D; ácidos aldónicos, ácidos sacáricos C07C 59/105, C07C 59/285; cianohidrinas C07C 255/16; glicales C07D; compuestos de constitución indeterminada C07G; polisacáridos, sus derivados C08B; ADN o ARN concerniente a la ingeniería genética, vectores, p. ej. plásmidos o su aislamiento, preparación o purificación C12N 15/00; industria del azúcar C13). › C07H 15/00 Compuestos que contienen radicales hidrocarbonados o hidrocarbonados sustituidos, unidos directamente a los heteroátomos de los radicales sacárido. › Sistemas cíclicos condensados que tienen tres o más ciclos.
PDF original: ES-2422290_T3.pdf
Fragmento de la descripción:
Procedimiento para preparar un producto intermedio sintético para la preparación de nucleósidos ramificados Esta invención se refiere a métodos para la preparación de un producto intermedio sintético en un procedimiento para la preparación de análogos de 2'-C-alquil-2'-halo-nucleósidos, que son importantes como agentes, anticancerosos, antivirales y antibacterianos.
Un procedimiento para la preparación de un producto intermedio de ribonolactona sustituida con halógeno que es útil en la síntesis de un análogo de 2'-C-metil-2'-halo-nucleósido sigue presentando retos, en particular cuando el átomo de halógeno es flúor.
Un producto intermedio clave en la preparación de análogos de azúcar utilizados en la síntesis de nucleósidos y vitaminas es la 2-C-metil-D-ribono-lactona. Ya en 1880 Scheibler describió un procedimiento para la preparación de la lactona (John Sowden, "The saccharinic acids” en Adv. Carbohydrate Chem. 12: 43-46 (1957) , citando a C. Scheibler, Berichte 13: 2212 (1880) ) . Desgraciadamente, los rendimientos del producto eran sólo aproximadamente un 10% (Id.) . Aproximadamente en el mismo tiempo, H. Kiliani sintetizó 2-metil-D-ribonolactona tratando D-fructosa con hidróxido de calcio (H. Kiliani, Berichte, 15: 2953 (1882) , como se cita en F. J. López-Herrera et al., J. Carbohydrate Chemistr y , 13 (5) : 767-775 (1994) ) . Sin embargo, el proceso requiere meses hasta llegar a la terminación y el rendimiento de producto era también sólo aproximadamente el 10% (Id. en 768) . Sin embargo el proceso de Kiliani le permitió establecer las posiciones de importantes grupos funcionales en el compuesto (John Sowden, " The saccharinic acids” en Adv. Carbohydrate Chem. 12: 43-46 (1957) , citando a H. Kiliani, Ann ., 213:361 (1883) ) .
A principios de la década de los 60, Whistler y BeMiller intentaron mejorar la síntesis de Kiliani (Roy L. Whistler y J.
N. BeMiller, "a-D-Glucosaccharino-1, 4-lactona" en Methods in Carbohydrate Chemistr y , 2: 484-485 (1963) ) . Whistler y BeMiller añadieron agua hirviendo e hidróxido de calcio a la D-fructosa, purgaron el sistema con gas nitrógeno, y repitieron el mismo proceso. La mezcla se mantuvo después durante 6 a 8 semanas, después de lo cual se trató con CO2 y ácido oxálico dihidrato, y se filtró bajo presión. El residuo se lavó repetidamente hasta una consistencia similar a la de un jarabe, y se reunieron los filtrados; el disolvente se evaporó bajo presión reducida y el producto resultante se dejó cristalizar con refrigeración. El rendimiento del producto final era todavía de sólo aproximadamente el 10% (Id. en 485) y el proceso precisó dos meses para completarse.
Los documentos BE 731271 y GB 1189973, cedidos a Deutsche Akademie der Wissenchaften, describieron un procedimiento para la preparación de 3'-fluoronucleósidos mediante la reacción de un nucleósido con un agente de fluoración tal como HF en un disolvente orgánico como el THF, a temperaturas en el intervalo de 130 a 160 °C.
En un intento de mejorar los rendimientos del producto, López-Aparicio et al. publicaron la síntesis de 2-C-metil-Dribono-1, 4-lactona a partir de 2, 3-O-isopropiliden-D-gliceraldehído como una alternativa a la síntesis de Kiliani (López-Aparicio et al., Carbohydrate Res., 129:99 (1984) , como se cita en F. J. Lopez Herrera et al., J. Carbohydrate Chemistr y , 13 (5) : 767-775 (1994) en 768-769. El proceso de López-Aparicio incluía condensar 2, 3-O-isopropilidenD-gliceraldehído con (1-metoxi-carbonil-etilideno) trifenilfosforano para producir E- (S) -4, 5-dihidroxi-4, 5-Oisopropiliden-2-metil-2-pentenoato de metilo; hidrolizar (en HCl) e isomerizar fotoquímicamente el pentenoato; lactonizar el producto de pentenoato para producir un butenólido; tritilar el butenólido en C-5 por reacción con cloruro de tritilo y piridina, a continuación realizar la cis-hidroxilación con permanganato potásico y cloruro de metileno en presencia de un éter corona. La eliminación final del grupo de tritilo (trifenilmetilo) se logró por la reacción con TFA (ácido trifluoroacético) (Id en 768) . López-Aparicio et al. publicaron rendimientos de producto de ribonolactona en aproximadamente el 80%, pero otros no pudieron reproducir esta cifra sobre la base de las cantidades de masa en gramos de los materiales proporcionados en la parte experimental de su publicación. En vez de ello, los cálculos indicaron un porcentaje de rendimiento de alrededor de 36% de ribonolactona. Además, el proceso de López-Aparicio et al. era mucho más complejo que la síntesis de Kiliani, requería el uso de reactivos tóxicos tales como el permanganato de potasio y de un equipo especializado para la irradiación para conseguir la isomerización fotoquímica, y tenía un tiempo de reacción mínimo de 60 horas (Id. en 768, 770-772) .
Ninguno de los planteamientos anteriores abordó el problema de la preparación de análogos de ribonucleósido 2'-Cramificados o 2'-disustituidos.
En 1989, la firma Asahi Glass Company Ltd. publicó la síntesis de fluoronucleósidos que tenían efectos antivirales y antitumorales (JP 02270864 y JP 01100190) . Estos nucleósidos fueron preparados mediante el tratamiento de un derivado de 9- (alfa-fluoro-4-beta-hidroxi-1-beta-ciclopentil) pirimidina con cloruro de trifluorometanosulfonilo, cloruro de p-toluenosulfonilo, cloruro de metanosulfonilo o cloruro de imidazolilsulfonilo en presencia de una base, seguido por reducción (JP 02270864) . En un segundo método de síntesis, se obtuvieron 2’, 3'-desoxi-2’, 3'-dideshidro-2'fluoronucleósidos mediante la deshidrogenación de un derivado 2'-desoxi-2'-fluororribofuranosilo, o mediante la deshidrogenación de un derivado 2', 3'-didesoxi-2'-fluoro-3'-halo-ribonucleósido (JP 01100190) .
En 1990, Bobek et al. describieron la síntesis de derivados antivirales, antitumorales y antimicrobianos de arabinopiranosil nucleósidos que tenían un átomo de flúor en la posición 2' del anillo de piranosa (patente de EE.UU. nº 4.918.056) . Estos compuestos fueron preparados mediante la condensación de una base de nucleótido pirimidina, purina, o 1, 3-oxazina con un 2-desoxi-2, 2-difluoro-D-arabinopiranósido acilado bloqueado con un grupo hidroxilo, y/o un 2-desoxi-2-bromo-2-fluoro-D-arabinopiranósido acilado (Id.) .
En 1997, Harr y -O'Kuru et al. describieron una ruta sintética para la preparación de ribonucleósidos 2'-C-ramificados (Harr y -O'Kuru et al., J. Org. Chem., 62: 1754-9 (1997) ) . La 1, 3, 5-tri-O-benzoil-a-D-ribofuranosa disponible comercialmente se utilizó como material de partida, que se preparó a partir de D-ribosa o D-arabinosa (Darabinopiranosa) . El 1, 3, 5-tri-O-benzoil-a-D-ribofuranosa fue oxidada en los 2-OH libres con reactivo per y odinano de Dess-Martin, y produjo 1, 3, 5-tri-O-benzoil-2-ceto-ribofuranosa, así como su correspondiente hidrato. El producto deseado y el hidrato se agitaron con un exceso de MgSO4 y se dejó reposar durante la noche. La mezcla se filtró después y se concentró con el fin de producir un producto de cetona sustancialmente puro. El 2-cetoazúcar resultante se trató con MeMgBr/TiCl4 (o, alternativamente, con MeTiCI3, CH2=CHMgBr/CeCl3, o TMSC=CLi / CeCl3) , que produjo una mezcla anomérica del alquil-, alquenil-o alquinil-ribofuranósido1, 3, 5-tri-O-benzoil-2-sustituido deseado y sus isómeros transesterificados, alquil, alquenil o alquinil ribofuranósido a- y º-2, 3, 5-tri-O-benzoil-2sustituido en una relación próxima a 5:3 de producto deseado a formas isoméricas (Id. en 1755) . Los ribofuranósidos 2-alquilados fueron después convertidos en un único producto deseado, 1, 2, 3, 5-tetrabenzoil-2-alquilrribofuranósido, mediante el tratamiento con cloruro de benzoílo, DMAP y trietilamina con un rendimiento declarado de aproximadamente 70% con una relación º/a de 4:1 (Id.) .
En 1998, Chambers et al. publicaron la síntesis de compuestos 2', 3'-didesoxi-3'-fluorouridina, mediante la reacción de un correspondiente anhidronucleósido con fluoruro de hidrógeno en presencia de un compuesto de organo-hierro y en un disolvente orgánico (patente de EE.UU. nº 5.717.086) .
Se han dado a conocer publicaciones recientes de síntesis de 2 ' y/o 3' halonucleósidos por Pharmasset, Inc., The University of Georgia Research Foundation, Inc., y la Universidad de Emor y .
El documento WO 05/003147 (también US 2005/0009737) de Pharmasset, Inc., describió la síntesis de análogos de 2'-C-metil-2'-fluoro nucleósido por una de dos rutas sintéticas generales: alquilación de un compuesto de hidrato de carbono apropiadamente modificado, fluoración y, a continuación acoplamiento a una base de nucleósido deseada,
o glicosilación de una base de nucleósido deseada para formar un nucleósido, a continuación... [Seguir leyendo]
Reivindicaciones:
1. Un procedimiento para preparar un compuesto de Fórmula II
en la que R1 es alquilo de cadena lineal o de cadena ramificada, cicloalquilo, alquenilo, alquinilo, alenilo, CF3, ciano, arilo, aralquilo, o heterocíclico, que comprende a) proporcionar un compuesto de estructura (i) o (ii)
en donde OR es un grupo lábil y X es halógeno;
b) poner en contacto el compuesto con un agente de fluoración bajo condiciones anhidras; y 10 c) eliminar el grupo protector de hidroxilo del producto de la etapa b) .
2. El procedimiento según la reivindicación 1ª, en el que R1 es metilo, etilo, vinilo o -C=CH.
3. El procedimiento según la reivindicación 1ª, en el que OR se elige entre el grupo que consiste en triflato, mesilato y tosilato.
4. El procedimiento según la reivindicación 1ª, en el que X en la Fórmula (II) es bromo, cloro o yodo.
5. El procedimiento según la reivindicación 1ª, en el que la reacción contiene menos de 1% de agua, menos de 0, 1% de agua, o menos de 0, 01% de agua.
6. El procedimiento según la reivindicación 1ª, que comprende además convertir un compuesto de Fórmula (II) en un compuesto de 1, 4-lactona.
7. El procedimiento según la reivindicación 6ª, que comprende: c) poner en contacto el producto de la etapa 20 (b) con un ácido orgánico en un disolvente orgánico.
8. El procedimiento según la reivindicación 7ª, en el que el ácido es ácido trifluoroacético.
9. El procedimiento según la reivindicación 7ª, en el que el ácido es un ácido aril o alquil sulfónico.
10. El procedimiento según la reivindicación 9ª, en el que el ácido es ácido metanosulfónico, o ácido ptoluenosulfónico.
11. El procedimiento según la reivindicación 7ª, en el que el disolvente es 1, 4-dioxano.
12. El procedimiento según la reivindicación 6ª, en el que el compuesto de 1, 4-lactona es un compuesto de Fórmula (B)
en la que R1 es alquilo inferior C1-4, alquenilo, alquinilo o CF3.
13. El procedimiento según la reivindicación 6ª, en el que el compuesto de 1, 4-lactona es 2-desoxi-2-fluoro-2-Cmetil-D-ribono-1, 4-lactona.
14. El procedimiento según la reivindicación 12ª, en el que R1 es acetileno o vinilo.
15. El procedimiento según la reivindicación 1ª, que comprende además: d) reducir el producto de 1, 5-lactona de la etapa c) para dar un hemiacetal; e) derivatizar el hidroxilo de la etapa d) para dar un grupo lábil adecuado; y f) poner en contacto el producto de 1, 4-lactona de la etapa e) con una base de purina o pirimidina para proporcionar un compuesto de nucleósido 2’-ramificado.
16. El procedimiento según la reivindicación 1ª, en el que el compuesto de Fórmula (i) es 3, 4-O-isopropiliden-2C-metil-2-O-tifluorometanosulfonil-D-arabinono-1, 5-lactona.
17. El procedimiento según la reivindicación 1ª, en el que se produce una 2-desoxi-2-fluoro-3, 4-O-isopropiliden2-C-metil-D-ribono-1, 5-lactona.
18. El procedimiento según la reivindicación 15ª, en el que se produce una 2-desoxi-2-fluoro-2-C-metil-Dribono-1, 4-lactona.
19. El procedimiento según la reivindicación 1ª, en el que la etapa b) se lleva a cabo a una temperatura por debajo de 0 °C, o a una temperatura por debajo de -5 °C.
20. Un procedimiento para preparar un compuesto 2-desoxi-2-alquil-2-fluoro-lactona, que comprende
a) hacer reaccionar un compuesto de isopropiliden, arabinono alquil-sustituido-1, 5-lactona para proporcionar una 2-desoxi-2- alquil-disustituido isopropiliden arabinono -1, 5-lactona sustituida con un grupo lábil en posición 2;
b) hacer reaccionar el producto procedente de la etapa a) con un agente de fluoración para proporcionar una 2-desoxi-2-fluoro, alquil-disustituido isopropiliden ribono-1, 5-lactona;
c) desproteger el producto procedente de la etapa b) para proporcionar una 2-desoxi-2-fluoro, alquildisustituido ribono-1, 4-lactona como producto mayoritario y una 2-desoxi-2-fluoro, alquil-disustituido ribono-1, 5lactona como producto minoritario; y
d) opcionalmente proteger uno o más grupos hidroxi en uno de los productos procedentes de la etapa c) .
21. El procedimiento según la reivindicación 20ª, etapa a) en donde la 2-desoxi-2-alquil-disustituido isopropiliden arabinono -1, 5-lactona sustituida con un grupo lábil en posición 2 es 3, 4-O-isopropiliden-2-C-metil-2-O-trifluorometanosulfonil-D-arabinono-1, 5-lactona.
22. El procedimiento según la reivindicación 20ª, etapa b) en el que la 2-desoxi-2-fluoro, alquil-disustituido isopropiliden ribono-1, 5-lactona 2 es 2-desoxi-2-fluoro-3, 4-O-isopropiliden-2-C-metil-D-ribono-1, 5-lactona.
23. El procedimiento según la reivindicación 20ª, etapa c) en donde el producto mayoritario 2-desoxi-2-fluoro, alquil-disustituido ribono-1, 4-lactona es 2-desoxi-2-fluoro-2-C-metil-D-ribono-1, 4-lactona, o la etapa c) en donde el producto minoritario 2-desoxi-2-fluoro, alquil-disustituido ribono-1, 5-lactona es 2-desoxi-2-fluoro-2-C-metil-D-ribono1, 5-lactona.
24. El procedimiento según la reivindicación 20ª, etapa d) en donde el benzoílo es el grupo protector en uno o más grupos hidroxi en cualquiera de los productos de la reivindicación 1ª, etapa c) .
25. El procedimiento según la reivindicación 1ª o la reivindicación 20ª, en el que el agente de fluoración es tris (dimetilamino) sulfonio difluorometil silicato (TASF) .
26. El procedimiento según la reivindicación 20ª, etapa c) en donde el reactivo desprotector es ácido trifluoroacético (TFA) .
27. El procedimiento según la reivindicación 20ª, etapa a) en donde el grupo lábil se elige entre el grupo consistente en triflato, mesilato, bromo, cloro o yodo.
28. El procedimiento según la reivindicación 27ª, en el que el grupo lábil es triflato.
Patentes similares o relacionadas:
Composición de Estevia, del 11 de Octubre de 2019, de Purecircle Usa Inc: Un proceso para la producción de una composición de estevia que comprende rebaudiósido B, que comprende las etapas de: proporcionar un edulcorante […]
Un edulcorante no calórico, del 31 de Julio de 2019, de Conagen Inc: Un rebaudiósido sintético de fórmula (I):**Fórmula**
Preparación de (S,S)-secoisolariciresinol diglucósido y (R,R)-secoisolariciresinol diglucósido, del 28 de Noviembre de 2018, de THE TRUSTEES OF THE UNIVERSITY OF PENNSYLVANIA: Un proceso para preparar un compuesto de fórmula ((S,S)-SDG-1) o un compuesto de fórmula (R,R)- SDG-2**Fórmula** comprendiendo el proceso: (a) […]
Purificación de epidaunorubicina, del 19 de Octubre de 2018, de MEDAC GESELLSCHAFT FUR KLINISCHE SPEZIALPRAPARATE MBH: Procedimiento para la purificación de epidaunorubicina que comprende las siguientes etapas: a) proporcionar una mezcla que contiene epidaunorubicina, epi-feudomicina […]
Isómeros del glucósido esteviol, del 13 de Julio de 2016, de PEPSICO, INC.: Un compuesto de fórmula II:**Fórmula** en la que R1 es hidrógeno y R2 es 2-(1-ß-D-glucopiranosil)-1-ß-D-glucopiranosilo, 2,3-bis(1-ß-D-glucopiranosil)1- ß-D-glucopiranosilo, […]
Derivados de oligosacáridos sulfatados novedosos, del 27 de Abril de 2016, de Progen PG500 Series Pty Ltd: Un compuesto de la fórmula general:**Fórmula** donde: X e Y son cada uno una unidad monosacárida donde cada grupo hidroxilo no comprendido en un enlace glucosídico es […]
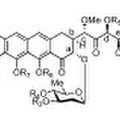 Derivados glicosilados de mitramicina, su procedimiento de obtención y sus usos, del 23 de Abril de 2014, de UNIVERSITY OF KENTUCKY RESEARCH FOUNDATION: La presente invención proporciona compuestos caracterizados por la fórmula (I), donde cada uno de los radicales sustituyente se describe en la memoria. La invención también […]
Derivados glicosilados de mitramicina, su procedimiento de obtención y sus usos, del 23 de Abril de 2014, de UNIVERSITY OF KENTUCKY RESEARCH FOUNDATION: La presente invención proporciona compuestos caracterizados por la fórmula (I), donde cada uno de los radicales sustituyente se describe en la memoria. La invención también […]
Derivados de ácidos aureólicos, su procedimiento de obtención y sus usos, del 19 de Marzo de 2014, de ENTRECHEM, S.L: Derivados de ácidos aureólicos, su procedimiento de obtención y sus usos. La presente invención proporciona una cepa bacteriana que produce compuestos pertenecientes […]