NUEVA SINTESIS DE COMPUESTOS INTERMEDIARIOS HETEROARILAMINA.
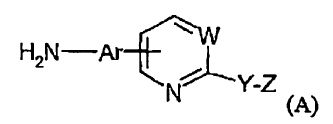
Procedimiento para preparar un compuesto de fórmula (A):
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/US01/15665.
Solicitante: BOEHRINGER INGELHEIM PHARMACEUTICALS INC..
Nacionalidad solicitante: Estados Unidos de América.
Dirección: 900 RIDGEBURY ROAD,P.O. BOX 368RIDGEFIELD, CT 068.
Inventor/es: KAPADIA, SURESH, R., SONG, JINHUA, J., YEE,NATHAN K.
Fecha de Publicación: .
Fecha Concesión Europea: 24 de Marzo de 2010.
Clasificación Internacional de Patentes:
- C07D213/38 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07D COMPUESTOS HETEROCICLICOS (Compuestos macromoleculares C08). › C07D 213/00 Compuestos heterocíclicos que contienen ciclos de seis miembros, no condensados con otros ciclos, con un átomo de nitrógeno como el único heteroátomo del ciclo y tres o más enlaces dobles entre miembros cíclicos o entre miembros cíclicos y miembros no cíclicos. › que tienen solamente hidrógeno, o radicales hidrocarbonados, unidos al átomo de nitrógeno sustituyente.
- C07D213/61 C07D 213/00 […] › Atomos de halógeno o radicales nitro.
Clasificación PCT:
- C07D213/38 C07D 213/00 […] › que tienen solamente hidrógeno, o radicales hidrocarbonados, unidos al átomo de nitrógeno sustituyente.
- C07D213/61 C07D 213/00 […] › Atomos de halógeno o radicales nitro.
Clasificación antigua:
- C07D213/38 C07D 213/00 […] › que tienen solamente hidrógeno, o radicales hidrocarbonados, unidos al átomo de nitrógeno sustituyente.
- C07D213/61 C07D 213/00 […] › Atomos de halógeno o radicales nitro.
Fragmento de la descripción:
Nueva síntesis de compuestos intermediarios heteroarilamina.
Campo de la invención
La presente invención se refiere a la síntesis de compuestos intermediarios heteroarilamina.
Antecedentes de la invención
Se han descrito ureas sustituidas con arilo o con heteroarilo las cuales inhiben la producción de citoquinas. Estos inhibidores se han descrito como compuestos terapéuticos eficaces en enfermedades mediadas por citoquinas, entre ellas las enfermedades inflamatorias y las autoinmunológicas. Se informa de ejemplos de este tipo de compuestos en las patentes WO nº 99/23091 y nº 98/52558.
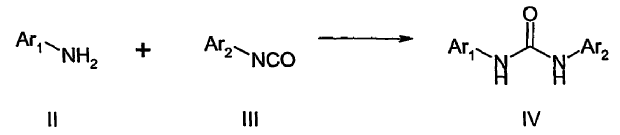
Una etapa crucial en la síntesis de dichos compuestos es la formación del enlace de urea. Se ha informado de diversos métodos para conseguirlo. Por ejemplo, tal como se informa en las referencias anteriormente indicadas, puede hacerse reaccionar una amina aromática o heteroaromática, II, con un isocianato aromático o heteroaromático, III, generando la urea, IV (Esquema I).
Esquema I
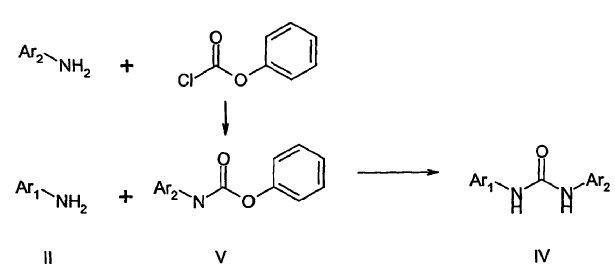
En el caso de que no se encuentre disponible comercialmente, puede prepararse el isocianato III mediante reacción de una arilamina o heteroarilamina, Ar2NH2, con fosgeno o con un equivalente de fosgeno, tal como carbonato de bis-triclorometilo (trifosgeno) (P. Majer y R.S. Randad, J. Org. Chem. 59:1937, 1994) o cloroformato de triclorometilo (difosgeno) (K. Kurita, T. Matsumura y Y. Iwakura, J. Org. Chem. 41:2070, 1976), formando el isocianato, III, seguido de la reacción con Ar1NH2 para proporcionar la urea. Entre otros enfoques para formar la urea que se han informado en la literatura química se incluyen la reacción de un carbamato con una arilamina o heteroarilamina (ver, por ejemplo, B. Thavonekham, Synthesis 1189, 1997, y T. Patonay et al., Synthetic Communications 26:4253, 1996), tal como se muestra en el Esquema II, a continuación, para un carbamato de fenilo. La solicitud de patente US nº 09/611.109 también da a conocer un procedimiento para preparar heteroarilureas mediante la reacción de intermediarios carbamato particulares con la arilamina deseada.
Esquema II
La solicitud de patente US nº 09/505.582 y la patente PCT nº US 00/03865 describen ureas inhibidoras de citoquinas, de fórmula (I).
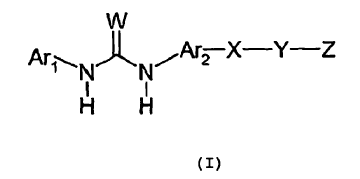
Una Ar2NH2 necesaria para preparar los compuestos preferentes indicados en las mismas se ilustra en la fórmula (A):

en la que W, Y y Z se definen posteriormente.
La síntesis de II, un intermediario preferente de fórmula (A), ha sido descrita en la solicitud de patente US nº 09/505.582 y en la patente PCT nº US 00/03865 y se ilustra en el Esquema III.
Esquema III
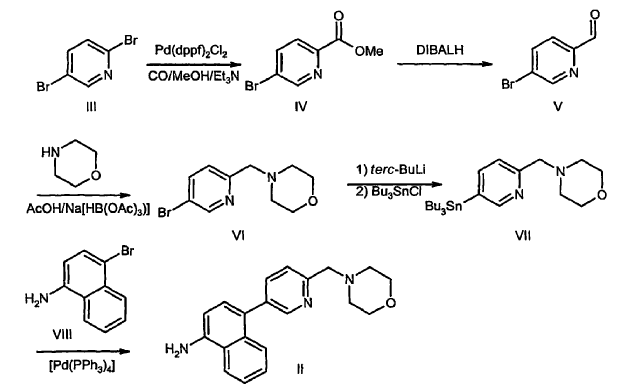
La síntesis se inicia con una carbonilación catalizada por paladio de la 2,5-dibromopiridina (III), proporcionando el éster IV con un rendimiento de 55%. La reacción se lleva a cabo bajo presión (80 psi de CO) y debe realizarse un seguimiento de la misma para minimizar la formación del diéster, un producto secundario no deseado. La reducción de IV con hidruro de diisobutilaluminio a -78ºC proporciona el aldehído V. Seguidamente se realiza una aminación reductora, proporcionando VI.
A continuación, se convierte el intermediario VI en II mediante la reacción con t-BuLi a -78ºC, seguido de cloruro de tributilestaño, proporcionando tributilestanano VII, seguido de acoplamiento de Stille catalizado por paladio con el intermediario VIII, que proporciona II. La conversión de VI e intermediarios análogos en otros intermediarios de fórmula II mediante acoplamiento de Suzuki también se describe en la solicitud de patente US nº 09/505.582 y en la patente PCT nº US 00/03865 (Esquema IV). Según este método, el intermediario IX se trata con n-BuLi, seguido de trimetilborato, proporcionando ácido arilborónico, X. El acoplamiento de Suzuki catalizado por paladio con VI proporciona XI, que se desprotege mediante tratamiento con ácido, proporcionando II.
Esquema 4
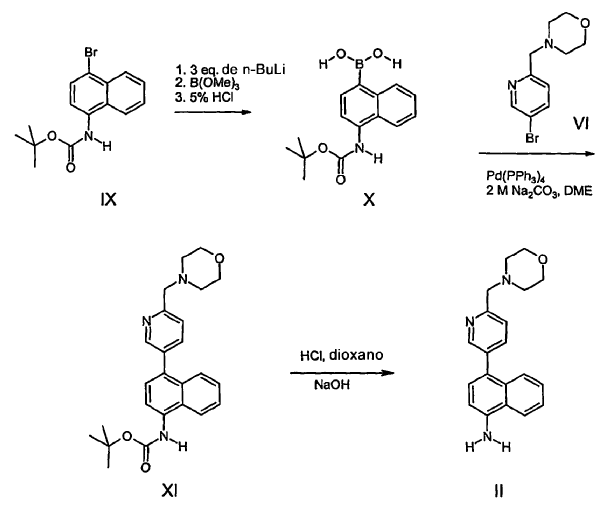
Este procedimiento no resulta adecuado para la utilización a gran escala y a escala comercial, por varios motivos. Una reacción (Esquema III) se lleva a cabo bajo presión elevada (80 psi) y otra, a temperatura extrema (-78ºC). El rendimiento de IV sólo es moderado y la formación de producto secundario exige una etapa de purificación. Dichos factores, más el coste de los materiales iniciales y los reactivos implica que este procedimiento resulta excesivamente caro a escala comercial.
La preparación de 2-bromo-5-litiopiridina mediante la reacción de 2,5-dibromopiridina con n-BuLi a -100ºC ha sido descrita anteriormente (W.E Parham y R.M. Piccirilli, J. Org. Chem. 42:257, 1977). La formación selectiva de cloruro de 2-bromo-5-piridinmagnesio mediante la reacción con 2,5-dibrompiridina con iPrMgCl a una temperatura de entre 0ºC y la temperatura ambiente también ha sido informada anteriormente (F. Trecourt et al., Tetrahedron Let. 40:4339, 1999). En estos casos, el intercambio entre metal y halógeno se produce exclusivamente en la posición 5 del anillo piridina. Sin embargo, las síntesis de cloruro de 5-bromo-2-piridinmagnesio y de cloruro de 5-cloro-2-piridinmagnesio no han sido informadas anteriormente.
La preparación de un intermediario de litio, 5-cloro-2-litiopiridina, a partir de 2-bromo-5-cloropiridina, ha sido informada anteriormente (U. Lehmann et al., Chem. Euro. J. 5:854, 1999). Si embargo, esta síntesis requiere la reacción con n-BuLi a -78ºC. La preparación de 5-bromo-2-litiopiridina a partir de 2,5-bromopiridina ha sido informada por X. Wang et al. (Tetrahedron Letters, 4335, 2000). Sin embargo, el método requiere condiciones criogénicas y de elevada dilución. La selectividad también depende del tiempo de reacción. No resulta adecuado para la síntesis a gran escala.
La síntesis del intermediario 5-bromo-2-yodopiridina mediante reflujo de 2,5-dibromopiridina en HI ha sido informada anteriormente (U. Lehmann, ibid.). Ya ha sido descrito un procedimiento en condiciones más suaves para la preparación de 2-yodopiridina a partir de 2-cloro o de 2-bromopiridina (R.C. Corcoran y S.H. Bang, Tetrahedron Lett. 31:6757, 1990).
Descripción resumida de la invención
Es un objetivo de la invención proporcionar: nuevos haluros de 2-(5-halopiridil)magnesio y de 2-(5-halopirimidinil)magnesio, nuevos métodos para la producción de los mismos, y un nuevo método de utilización de dichos haluros en la síntesis eficiente de sus piridinas y pirimidinas 5-halo-2-sustituidas.
También es un objetivo de la invención proporcionar un nuevo método de producción de heteroarilaminas de fórmula (A):
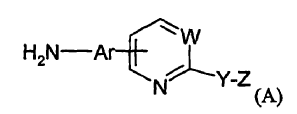
en la que Ar, W, Y y Z se definen posteriormente; las heteroarilaminas resultan útiles en la producción de heteroarilureas, tal como se ha indicado anteriormente.
Descripción detallada de las realizaciones preferentes
La presente invención se refiere a una nueva estrategia para la síntesis de compuestos heteroarilamina de fórmula (A) que constituyen el componente crucial de compuestos farmacéuticamente activos que presentan un grupo heteroarilurea.
Por lo tanto, la invención proporciona procedimientos de preparación de un compuesto de fórmula (A):
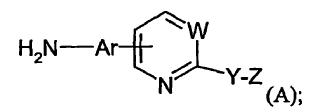
en la que:
W es CR3 o N, en donde R3 se selecciona de entre hidrógeno, alquilo C1-5, alcoxi C1-5, arilalquilo C0-5 y -COR4, en donde R4 se selecciona...
Reivindicaciones:
1. Procedimiento para preparar un compuesto de fórmula (A):
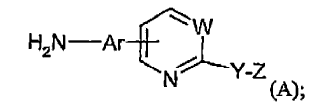
en la que:
W es CR3 o N, en donde R3 se selecciona de entre hidrógeno, alquilo C1-5, alcoxi C1-5, aril-C0-5-alquilo y -COR4, en el que R4 se selecciona de entre alquilo C1-5, alcoxi C1-5, aril-C0-5-alquilo y amino que opcionalmente se monosustituye o disustituye con alquilo C1-5 y aril-C0-5-alquilo,
Ar se selecciona de entre:
fenilo, naftilo, quinolinilo, isoquinolinilo, tetrahidronaftilo, tetrahidroquinolinilo, tetrahidroisoquinolinilo, bencimidazolilo, benzofuranilo, dihidrobenzofuranilo, indolinilo, benzotienilo, dihidrobenzotienilo, indanilo, indenilo e indolilo, opcionalmente sustituyendo cada uno con uno o más R1 o R2,
Y se selecciona de entre:
un enlace, una cadena de carbonos C1-4 saturada o insaturada ramificada o no ramificada opcionalmente halogenada parcial o totalmente, en la que uno o más grupos metileno se reemplazan opcionalmente con O, N o S(O)m, y en en la que Y opcionalmente se sustituye independientemente con uno a dos grupos oxo, fenilo o uno o más alquilos C1-4 opcionalmente sustituidos con uno o más átomos de halógeno, en el que, en el caso de que Y sea la cadena de carbonos, el átomo terminal del lado izquierdo de Y es un carbono,
Z se selecciona de entre:
arilo, heteroarilo seleccionado de entre piridinilo, piperazinilo, pirimidinilo, piridazinilo, pirazinilo, imidazolilo, pirazolilo, triazolilo, furanilo, tienilo y piranilo, y heterociclo seleccionado de entre tetrahidropirimidonilo, ciclohexanolilo, 2-oxo ó 2-tio-5-aza-biciclo[2.2.1]heptanilo, sulfidilo de pentametileno, sulfoxidilo de pentametileno, sulfonilo de pentametileno, sulfidilo de tetrametileno, sulfoxidilo de tetrametileno o sulfonilo de tetrametileno, tetrahidropiranilo, tetrahidrofuranilo, 1,3-dioxolanonilo, 1,3-dioxanonilo, 1,4-dioxanilo, morfolinilo, tiomorfolinilo, tiomorfolinilsulfoxidilo, tiomorfolinilsulfonilo, piperidinilo, piperidinonilo, pirrolidinilo y dioxolanilo, cada una de las Z anteriormente indicadas se sustituye opcionalmente con uno a tres halógenos, alquilo C1-6, alcoxi C1-6, alcoxi C1-3-alquilo C1-3, alcoxicarbonilo C1-6, aroilo, acilo C1-3, oxo, piridinilalquilo C1-3, imidazolilalquilo C1-3, tetrahidrofuranilalquilo C1-3, nitrilo-alquilo C1-3, nitrilo, fenilo en el que el anillo fenilo se sustituye opcionalmente con uno a dos halógenos, alcoxi C1-6 o monoalquilamino C1-3 o dialquilamino C1-3, alquilo-S(O)m C1-6 o fenilo-S(O)m, en el que el anillo fenilo se sustituye opcionalmente con uno a dos halógenos, alcoxi C1-6, halógeno o monoalquilamino C1-3 o dialquilamino C1-3,
o Z se sustituye opcionalmente con uno a tres aminos o aminoalquilos C1-3, en los que el átomo de N opcionalmente se monosustituye o disustituye independientemente con aminoalquilo C1-6, alquilo C1-3, arilalquilo C0-3, alcoxi C1-5-alquilo C1-3, alcoxi C1-5, aroilo, acilo C1-3, alquilo-S(O)m C1-3- o arilalquilo-S(O)m-C0-3, en los que cada uno de los alquilo y arilo anteriormente indicados que se encuentran unidos al grupo amino se sustituye opcionalmente con uno a dos halógenos, alquilo C1-6 o alcoxi C1-6,
o Z se sustituye opcionalmente con uno a tres arilos, heterociclo o heteroarilo tal como se ha indicado anteriormente en el presente párrafo, opcionalmente sustituyendo cada uno, a su vez, con halógeno, alquilo C1-6 o alcoxi C1-6,
o Z es nitrilo, amino en el que el átomo de N opcionalmente se monosustituye o disustituye independientemente con alquilo C1-6 o alcoxi C1-3-alquilo C1-3, alquilo C1-6 ramificado o no ramificado, alcoxi C1-6, nitrilalquilo C1-4, alquilo-S(O)m C1-6, arilo seleccionado de entre fenilo, piridinilo, pirimidinilo, piridazinilo, pirazinilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, furanilo, tienilo y piranilo, sustituyendo opcionalmente cada arilo con uno a tres halógenos, alquilo C1-6, alcoxi C1-6, dialquilamino C1-3, alquilo-S(O)m C1-6 o nitrilo y fenil-S(O)m, en los que el anillo fenilo se sustituye opcionalmente con uno a dos halógenos, alcoxi C1-6 o monoalquilamino C1-3 o dialquilamino C1-3,
R1 y R2 se seleccionan independientemente de entre:
un alquilo C1-6 ramificado o no ramificado opcionalmente halogenado parcial o totalmente, acetilo, aroilo, alcoxi C1-4 ramificado o no ramificado, opcionalmente estando cada uno parcial o totalmente halogenado, halógeno, metoxicarbonilo, alquilo-S(O)m C1-3 opcionalmente halogenado parcial o totalmente, o fenilsulfonilo,
m=0, 1 ó 2,
comprendiendo dicho procedimiento:
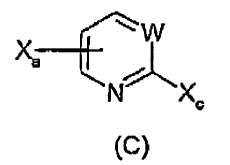
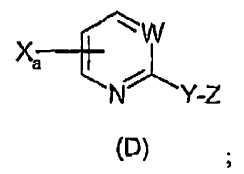
i) en un procedimiento de una etapa, hacer reaccionar un compuesto de fórmula (C) con un reactivo de Grignard, R-Mg-Bb, seguido de la adición de un compuesto E-Y-Z, en el que Y-Z es tal como se ha indicado anteriormente, estando adicionalmente caracterizado dicho componente E-Y-Z por ser un derivado electrofílico de Y-Z, teniendo lugar dicha reacción en un solvente aprótico adecuado a una temperatura de entre -78ºC y rt, durante un tiempo de reacción entre 1/2 hora y 2 horas, y aislando el compuesto de fórmula (D):
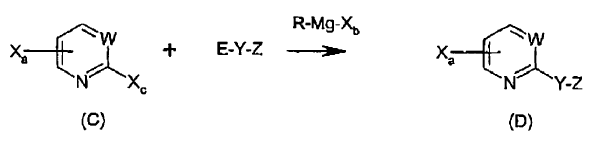
en la que:
Xa se selecciona de entre Br y Cl, y se encuentra unido al anillo en la posición 4 ó 5,
Xb se selecciona de entre Br, Cl e I,
Xc es I,
R se selecciona de entre arilo, alquilo C1-6 y cicloalquilo C5-7,
ii) hacer reaccionar el compuesto de fórmula (D) de la etapa a) con un ácido arilborónico de fórmula (E) en presencia de un catalizador seleccionado de entre níquel, paladio y una combinación de una fuente de paladio y un ligando apropiado, en un solvente adecuado a una temperatura de entre 0ºC y 150ºC durante aproximadamente 1 a 24 horas:
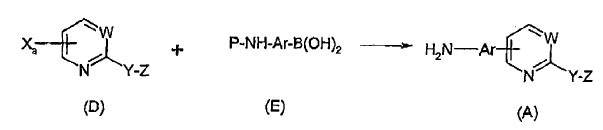
en donde P en la fórmula (E) es un grupo protector de amino, y posteriormente eliminar dicho grupo protector bajo condiciones adecuadas para producir un compuesto de fórmula (A).
2. Procedimiento según la reivindicación 1, en el que W es CH o N.
3. Procedimiento según la reivindicación 2, en el que:
en la etapa i):
Xa es Br unido en la posición 5 del anillo,
la temperatura de reacción es preferentemente de entre 0ºC y rt,
el tiempo de reacción es de 1 hora,
R es alquilo C1-6,
en la etapa ii):
el catalizador es un catalizador de paladio seleccionado de entre Pd(PPh3)2Cl2, Pd(PPh3)4, PdCl2(DPPE), PdCl2(DPPB), PdCl2(DPPP), PdCl2(DPPF) y Pd/C,
o el catalizador es una combinación de una fuente de paladio y un ligando apropiado, en donde la fuente de Pd se selecciona de entre PdCl2, Pd(OAc)2, Pd2(DBA)3 y Pd(DBA)2, y en donde
el ligando se selecciona de entre PPh3, DPPF, DPPP, DPPE, DPPB, P(o-tolilo)3, P(2,4,6-trimetoxifenilo)3, AsPh3, P(tBu)3 y BINAP,
la temperatura es de entre 25ºC y 100ºC,
el tiempo de reacción es de aproximadamente 15 horas,
el solvente se selecciona de entre DME, THF, tolueno, cloruro de metileno y agua.
4. Procedimiento según la reivindicación 3,
en la etapa i) R es isopropilo,
en la etapa ii):
el catalizador es una combinación de una fuente de paladio y un ligando apropiado, en donde la fuente de paladio es PdCl2 y el ligando es PPh3, y
el solvente es DME.
5. Procedimiento según la reivindicación 4, en el que:
W es CH,
Ar se selecciona de entre naftilo, quinolinilo, isoquinolinilo, tetrahidronatilo, tetrahidroquinolinilo, tetrahidroisoquinolinilo, indanilo, indenilo e indolilo, estando cada uno opcionalmente sustituido con uno o más grupos R1 o R2,
Y se selecciona de entre:
un enlace y
una cadena de carbonos C1-4 saturada o insaturada en la que uno de los átomos de carbono se reemplaza opcionalmente con O, N o S(O)m y en la que Y opcionalmente se sustituye independientemente con uno a dos grupos oxo, fenilo o uno o más alquilos C1-4 opcionalmente sustituidos con uno o más átomos de halógeno, en donde Y es la cadena de carbonos, el átomo terminal del lado izquierdo de Y es un carbono (el átomo que se encuentra unido covalentemente al heterociclo que presenta W).
Z se selecciona de entre:
fenilo, piridilo, pirimidinilo, piridazinilo, pirazinilo, imidazolilo, furanilo, tienilo, dihidrotiazolilo, dihidrotiazolilo, sulfoxidilo, piranilo, pirrolidinilo que se sustituyen opcionalmente con uno a tres nitrilos, alquilo C1-3, alcoxi C1-3, amino o monoalquilamino C1-3 o dialquilamino C1-3,
tetrahidropiranilo, tetrahidrofuranilo, 1,3-dioxolanonilo, 1,3-dioxanonilo, 1,4-dioxanilo, morfolinilo, tiomorfolinilo, tiomorfolinil-sulfoxidilo, pentametilensulfonilo, tetrametilen-sulfidilo, tetrametilen-sulfoxidilo o tetrametilen-sulfonilo que se encuentran opcionalmente sustituidos con uno a tres nitrilos, alquilo C1-3, alcoxi C1-3, amino o monoalquilamino C1-3 o dialquilamino C1-3, nitrilo, alquilo-S(O)m C1-3, halógeno, alcoxi C1-4, amino, monoalquilamino C1-6 o dialquilamino C1-6 y dialquilaminocarbonilo C1-3.
6. Procedimiento según la reivindicación 5, en el que:
Ar es naftilo,
Y se selecciona de entre:
un enlace y
una cadena de carbonos C1-4 saturada en la que el átomo terminal del lado izquierdo de Y es un carbono (el átomo que se encuentra unido covalentemente al heterociclo que presenta W) y uno de los otros átomos de carbono opcionalmente se reemplaza por O, N o S, y en la que Y opcionalmente se sustituye independientemente con un grupo oxo,
Z se selecciona de entre:
fenilo, piridinilo, pirimidinilo, piridazinilo, pirazinilo, imidazolilo, dihidrotiazolilo, sulfóxido de dihidrotiazolilo, piranilo y pirrolidinilo que opcionalmente se sustituyen con uno a dos alquilos C1-2 o alcoxi C1-2,
tetrahidropiranilo, morfolinilo, tiomorfolinilo, sulfoxidilo de tiomorfolinilo, piperidinilo, piperidinonilo, piperazinilo y tetrahidropirimidonilo que opcionalmente se sustituyen con uno a dos alquilos C1-2 o alcoxi C1-2,
y alcoxi C1-3.
7. Procedimiento según la reivindicación 6, en el que:
Ar es 1-naftilo, en el que NH2 se encuentra en la posición 4, y
Y se selecciona de entre:
un enlace, -CH2-, -CH2CH2- y -C(O)-.
8. Procedimiento según la reivindicación 7, en el que:
Y es -CH2-, y
Z es morfolinilo.
9. Procedimiento de preparación de un compuesto de fórmula (C) a partir de un compuesto de fórmula (B):
comprendiendo dicho procedimiento:
hacer reaccionar un compuesto de fórmula (B) con RxCOCl o (RxCO)2O y un yoduro metálico en un solvente adecuado a una temperatura de entre 25ºC y 150ºC, en donde Rx se selecciona de entre alquilo C1-7, -CF1-3 y -CCl1-3,

Xa se selecciona de entre Br y Cl, y Xa se encuentra unido en la posición 4 ó 5 del anillo,
Xoc es I,
W es CR3 o N, en donde R3 se selecciona de entre hidrógeno, alquilo C1-5, alcoxi C1-5, arilalquilo C0-5 y -COR4, en el que R4 se selecciona de entre alquilo C1-5, alcoxi C1-5, arilalquilo C0-5 y amino que opcionalmente se monosustituye o disustituye con alquilo C1-5 y arilalquilo C0-5, y
L es un grupo saliente adecuado.
10. Procedimiento según la reivindicación 9, en el que W es CH, CCH3 o N.
11. Procedimiento según la reivindicación 10, en el que:
X3 es Br unido en la posición 5 del anillo,
L se selecciona de entre Cl, Br, -OCORy y -OS(O)mRy, en el que Ry es arilo opcionalmente sustituido con alquilo C1-4 opcionalmente halogenado, o Ry es alquilo C1-4 opcionalmente halogenado; el metal se selecciona de entre Na y K,
y el solvente se selecciona de entre acetonitrilo, acetona, DMSO, DMF y THF.
12. Procedimiento según la reivindicación 11, en la que:
el compuesto de fórmula (B) se hace reaccionar con AcCl y NaI en acetonitrilo a una temperatura de entre 70ºC y 90ºC, y
L se selecciona de entre Br y Cl.
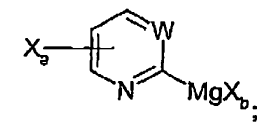
13. Procedimiento de preparación de un compuesto de Grignard de fórmula (F):
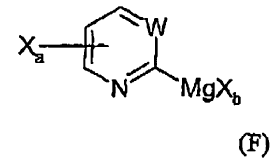
comprendiendo dicho procedimiento:
hacer reaccionar un compuesto de fórmula (C):
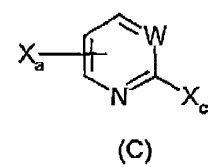
con un reactivo de magnesio de fórmula R-MgXb, teniendo lugar dicha reacción en un solvente aprótico adecuado a una temperatura de entre -78ºC y rt, durante un tiempo de reacción de entre 1/2 hora y 2 horas, produciendo el compuesto de Grignard de fórmula (F),
y en donde:
Xa se selecciona de entre Br y Cl, y Xa se une en la posición 4 ó 5 del anillo,
Xb se selecciona de entre Br, Cl e I,
Xc es I,
W es CR3 o N, en el que R3 se selecciona de entre hidrógeno, alquilo C1-5, alcoxi C1-5, arilalquilo C0-5 y -COR4, en el que R4 se selecciona de entre alquilo C1-5, alcoxi C1-5, arilalquilo C0-5 y amino que opcionalmente se monosustituye o disustituye independientemente con alquilo C1-5 y arilalquilo C0-5; W es preferentemente CH o N, y
R se selecciona de entre arilo, alquilo C1-5 y cicloalquilo C5-7.
14. Procedimiento según la reivindicación 13, en el que W es CH, CCH3 o N.
15. Procedimiento según la reivindicación 14, en el que:
W es CH,
Xa es Br y se encuentra unido en la posición 5 del anillo,
Xc es I,
la temperatura es de entre 0ºC y rt, y
el tiempo de reacción es de 1 hora.
16. Compuesto de fórmula:
Xa se selecciona de entre Br y Cl, y Xa se encuentra unido en la posición 4 ó 5 del anillo,
Xb se selecciona de entre Br, Cl e I, y
W es CR3 o N, en el que R3 se selecciona de entre hidrógeno, alquilo C1-5, alcoxi C1-5, arilalquilo C0-5 y -COR4, en el que R4 se selecciona de entre alquilo C1-5, alcoxi C1-5 y amino que opcionalmente se monosustituye o disustituye independientemente con alquilo C1-5 y arilalquilo C0-5.
17. Compuesto según la reivindicación 16, en el que W es CH, C-CH3 o N.
18. Compuesto según la reivindicación 17, en el que el compuesto se selecciona de entre:
19. Procedimiento de preparación de compuestos de fórmula (A) según la reivindicación 1,
y en la que, para la fórmula (A),
Y es -CH2-, y
Z se selecciona de entre:
heterociclo seleccionado de entre morfolino, tiomorfolino, piperidinilo y pirrolidinilo, cada Z anteriormente indicada puede sustituirse opcionalmente con uno a tres halógenos, alquilo C1-6, alcoxi C1-6, alcoxi C1-3-alquilo C1-3, alcoxicarbonilo C1-6, aroilo, acilo C1-3, piridinilalquilo C1-3, imidazolilalquilo C1-3, tetrahidrofuranilalquilo C1-3, nitrilalquilo C1-3, nitrilo, fenilo en el que el anillo fenilo se sustituye opcionalmente con uno a dos halógenos, alcoxi C1-6, dialquilamino C1-3, alquil-S(O)m- C1-6 o fenil-S(O)m-, en donde el anillo se sustituye opcionalmente con uno a dos halógenos, alcoxi C1-6 o dialquilamino C1-3,
o Z se sustituye opcionalmente con uno a tres aminos o aminoalquilo C1-3, en el que el átomo de N opcionalmente se disustituye independientemente con aminoalquilo C1-3, alquilo C1-3, arilalquilo C0-3, alcoxi-C1-5-alquilo C1-3, alcoxi C1-5, aroilo, acilo C1-3, alquil-S(O)m-C1-3 o arilalquil-S(O)m-C0-3; cada uno de los alquilo y arilo anteriormente indicados unidos al grupo amino se sustituye opcionalmente con uno a dos halógenos, alquilo C1-6 o alcoxi C1-6,
o Z se sustituye opcionalmente con uno a tres arilos o heterociclos, tal como se ha indicado anteriormente en la presente memoria en el presente párrafo, opcionalmente sustituyendo cada uno, a su vez, con halógeno, alquilo C1-6 o alcoxi C1-6,
o Z es amino, en el que el átomo de N opcionalmente se monosustituye o disustituye independientemente con alquilo C1-6 o alcoxi-C1-3-alquilo C1-3, alquilo C1-6 ramificado o no ramificado, alcoxi C1-6, nitrilalquilo C1-4, alquil-S(O)m- C1-6, arilo seleccionado de entre fenilo, piridinilo, pirimidinilo, piridazinilo, pirazinilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, furanilo, tienilo y piranilo, opcionalmente sustituyendo cada arilo con uno a tres halógenos, alquilo C1-6, alcoxi C1-6, dialquilamino C1-3, alquil-S(O)m-C1-6 o nitrilo, y fenil-S(O)m, en el que el anillo fenilo se sustituye opcionalmente con uno a dos halógenos, alcoxi C1-6 o monoalquilamino C1-3 o dialquilamino C1-3,
comprendiendo dicha reacción:
i) hacer reaccionar un compuesto de fórmula (C):
con un reactivo de magnesio de fórmula R-MgXb, teniendo lugar dicha reacción en un solvente aprótico adecuado a una temperatura de entre -78ºC y rt, durante un tiempo de reacción entre 1/2 hora y 2 horas, produciendo el compuesto de Grignard (F):
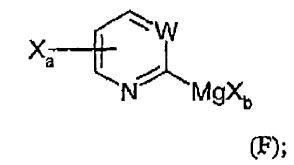
en la que:
Xa se selecciona de entre Br y Cl, y Xa se une al anillo en la posición 4 ó 5,
Xb se selecciona de entre Br, Cl e I,
Xc es I, y
R se selecciona de entre arilo, alquilo C1-6 y cicloalquilo C5-7,
ii) hacer reaccionar seguidamente el compuesto de Grignard (F) de la etapa anterior con una N,N-dialquilformamida, formando un aldehído, y aislar el aldehído,
iii) hacer reaccionar el aldehído de la etapa ii) con un grupo Z apropiado bajo condiciones ácidas en un solvente adecuado, proporcionando el compuesto (D):
haciendo reaccionar el compuesto de fórmula (D) con un ácido arilborónico de fórmula (E) y un catalizador de paladio, en presencia de un ligando apropiado, en un solvente adecuado a una temperatura de entre 0ºC y 150ºC, durante aproximadamente 1 a 24 horas:
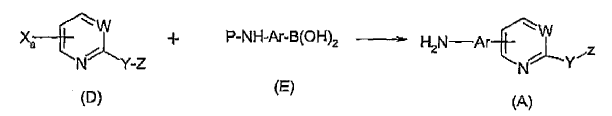
en la que P en la fórmula (E) es un grupo protector de amino, y posteriormente eliminar dicho grupo protector bajo condiciones adecuadas para producir un compuesto de fórmula (A).
20. Procedimiento según la reivindicación 19, en el que W es CH o N.
21. Procedimiento según la reivindicación 20, en el que:
en la etapa i):
Xa es Br y unido en la posición 5 del anillo,
W es CH,
R es alquilo C1-6,
en la etapa ii):
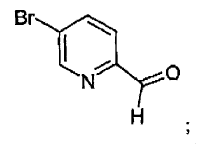
la N,N-dialquilformamida es DMF,
el aldehído es:
en la etapa iii):
las condiciones ácidas utilizan un ácido seleccionado de entre HCl, AcOH y H2SO4,
el solvente se selecciona de entre THF, cloruro de metileno, 1,2-dicloroetano; el tiempo es de aproximadamente 2 horas a aproximadamente la temperatura ambiente, seguido de la reacción in situ durante aproximadamente 2 horas.
22. Procedimiento según la reivindicación 21, en el que:
en la etapa i), R es isopropilo, y
en la etapa iii), el ácido es AcOH, y el solvente es 1,2-dicloroetano.
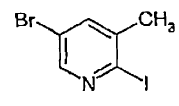
23. Compuesto de fórmula:
Patentes similares o relacionadas:
Derivados de heteroarilo y sus usos, del 25 de Septiembre de 2019, de MMV Medicines for Malaria Venture: Un compuesto de la Fórmula I:**Fórmula** en el que: R1 es piridilo, pirimidinilo o piracinilo, unido a través de un átomo de carbono del anillo heteroarilo, […]
Heterociclos capaces de modular las respuestas de linfocitos T, y procedimientos de uso de los mismos, del 27 de Febrero de 2019, de Nogra Pharma Limited: Un compuesto heterocíclico seleccionado del grupo que consiste en:**Fórmula** y**Fórmula** o una sal farmacéuticamente aceptable del mismo.
Inhibidores marcados del antígeno de membrana específico de la próstata (PSMA), evaluación biológica y uso como agentes para la obtención de imágenes, del 14 de Febrero de 2019, de THE JOHNS HOPKINS UNIVERSITY: Un compuesto de fórmula VII: **(Ver fórmula)** en la que: R" es H o alquilo C1-C6; Rx es arilo sustituido con: **(Ver fórmula)** Ry es H o alquilo C1-C6; […]
Inhibidor de la producción de melanina, del 16 de Enero de 2019, de POLA CHEMICAL INDUSTRIES, INC.: El uso no terapéutico de un compuesto representado por la siguiente fórmula general (excluyendo el clotrimazol) y/o una sal farmacológicamente aceptable del mismo […]
Heterodímeros de ácido glutámico, del 2 de Octubre de 2018, de MOLECULAR INSIGHT PHARMACEUTICALS, INC: Un compuesto o una sal farmacéuticamente aceptable del mismo, que comprende un resto 5 de unión a PSMA de glutamato-urea-lisina de Fórmula (I):**Fórmula** en el que el […]
Reactivos con funcionalidad tiol y sus usos, del 4 de Abril de 2018, de Glythera Limited: Un método para modificar un polipéptido que al menos tiene un grupo tiol reactivo, método que comprende poner en contacto el polipéptido con un reactivo […]
Inhibidores de proteasa, del 5 de Abril de 2017, de TaiMed Biologics, Inc. (100.0%): Un compuesto de fórmula I **Fórmula** o una sal farmacéuticamente aceptable del mismo, en donde Cx se selecciona del grupo que consiste […]
Procedimiento para preparar industrialmente amino-5,6,7,8-tetrahidronaftol sustituido con nitrógeno, del 1 de Marzo de 2017, de Shan Dong Luye Pharmaceutical Co., Ltd: Un procedimiento para preparar un compuesto de fórmula (I), que comprende:**Fórmula** hacer reaccionar un compuesto de fórmula (II) con un compuesto de fórmula (III) […]