CIP-2021 : A61K 9/22 : del tipo de liberación prolongada o discontinua.
CIP-2021 › A › A61 › A61K › A61K 9/00 › A61K 9/22[2] › del tipo de liberación prolongada o discontinua.
Notas[t] desde A61 hasta A63: SALUD; SALVAMENTO; DIVERSIONES
A NECESIDADES CORRIENTES DE LA VIDA.
A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE.
A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D).
A61K 9/00 Preparaciones medicinales caracterizadas por un aspecto particular.
A61K 9/22 · · del tipo de liberación prolongada o discontinua.
CIP2021: Invenciones publicadas en esta sección.
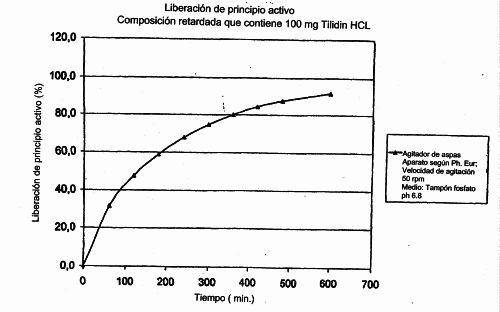
COMPOSICION FARMACEUTICA SOLIDA QUE CONTIENE CLORHIDRATO DE TILIDINA.
(16/02/2007). Ver ilustración. Solicitante/s: STADA ARZNEIMITTEL AG. Inventor/es: SCHUMANN, CHRISTOF, RENZ, JESSICA.
Composición farmacéutica, estable, sólida para la administración oral, que incluye clorhidrato de tilidina o bien uno de sus hidratos, un antagonista de morfina, así como sustancias auxiliares farmacéuticas, con la reserva de que se excluyen aquellas composiciones que contienen formadores de complejos para el ácido pirazolacético o cationes metálicos de valencia 2 ó 3, que se caracteriza por que contiene al menos un retardador, elegido entre las alquilcelulosas, alginatos, copolímeros de ácido acrílico, copolímeros de ácido metacrílico, hidroxialquilcelulosas , polivinilpirrolidona , alcoholes grasos, alcoholes de polivinilo, ésteres de ácidos grasos glicerina y ceras.
COMPRIMIDO DISGREGABLE POR VIA ORAL QUE FORMA UNA SUSPENSION VISCOSA.
(01/02/2007). Solicitante/s: CIMA LABS INC.. Inventor/es: SIEBERT, JOHN, M., KHANKARI, RAJENDRA, K., KOSITPRAPA, UNCHALEE, PATHER, S., INDIRAN.
Una forma de dosificación que se disgrega en la boca de un paciente que comprende: al menos un ingrediente activo en forma de un polvo o microcápsula y en una cantidad que es suficiente para provocar una respuesta terapéutica; una carga; y al menos un material potenciador de la viscosidad en la boca en una cantidad que es eficaz para proporcionar una suspensión viscosa organolépticamente aceptable después de la disgregación de la forma de dosificación en la boca de un paciente, en la que la viscosidad de dicha suspensión varía entre 25 y 500 Pascales Segundo (25.000 y 500.000 CPS), y en la que dicha forma de dosificación comprende adicionalmente al menos un par efervescente.
GRANULO ESTABLE Y FACIL DE PROCESAR, EN FORMA DE AMLODIPINA MALEATO.
(16/12/2006). Solicitante/s: PHARMA PASS II LLC. Inventor/es: SETH, PAWAN.
Gránulo que comprende entre el 1 y el 15% en peso de gránulo de la sal de maleato de amlodipina y entre el 2 y el 30% en peso de gránulo de la polivinilpirrolidona y un portador inerte.
FORMA DE DOSIFICACION PARA SUMINISTRAR UNA DOSIS CRECIENTE.
(16/12/2006). Ver ilustración. Solicitante/s: ALZA CORPORATION. Inventor/es: HAMEL, LAWRENCE, G., AYER, ATUL, DEVDATT, WRIGHT, JERI, D., LAM, ANDREW, SHIVANAND, PADMAJA.
ESTA INVENCION SE REFIERE A UNA FORMA POSOLOGICA Y A UN PROCEDIMIENTO DESTINADOS A ADMINISTRAR EN UN SUJETO HUMANO UN MEDICAMENTO EN CANTIDADES CRECIENTES EN EL TIEMPO.
COMPOSICIONES MEDICINALES ESTABLES PARA ADMINISTRACION ORAL QUE UTILIZAN OXIDOS DE HIERRO.
(01/12/2006) Una composición farmacéutica estable para uso oral que es una preparación de liberación sostenida de tipo matriz y que comprende: (a) un fármaco; (b) de 5 a 80% en peso de una base hidrófila que tiene una solubilidad suficiente para que la cantidad de agua necesaria para disolver 1 g de dicha base hidrofila sea 5 ml o menos a 20 ± 5ºC; (c) de 10 a 95% en peso de óxido de polietileno (i) que tiene una viscosidad de 2.000 cP o más como solución acuosa al 2% a 25ºC o (ii) que tiene un peso molecular de viscosidad media de 2.000.000 o más, dispersándose dicho fármaco y base hidrófila en una matriz de dicho óxido de polietileno, y (d) óxido férrico amarillo y/o óxido férrico rojo (i) siendo la cantidad de dicho óxido férrico amarillo y/o dicho óxido férrico rojo…
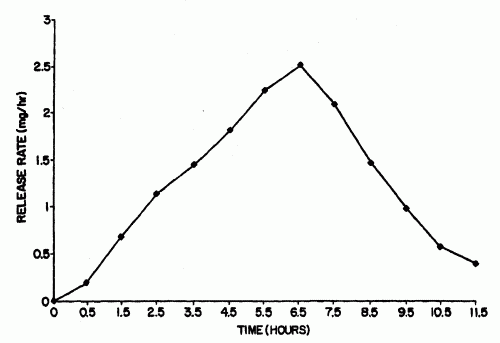
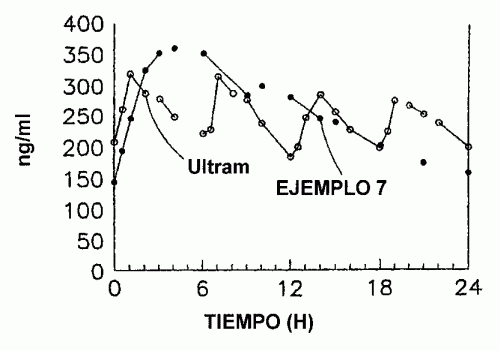
FORMULACIONES DE TRAMADOL DE LIBERACION SOSTENIDA ESTABILIZADA.
(01/12/2006). Ver ilustración. Solicitante/s: EURO-CELTIQUE S.A.. Inventor/es: OSHLACK, BENJAMIN, CHASIN, MARK, GOLDENHEIM, PAUL, HUANG, HUA-PIN.
Una forma de dosificación sólida oral de liberación sostenida estabilizada que contiene tramadol como el agente activo, que comprende una cantidad eficaz de tramadol o una sal farmacéuticamente aceptable del mismo dispersa en una matriz de un material hidrófobo que comprende una sustancia de tipo cera que se fundió o ablandó durante la preparación de dicha matriz, estando dicha forma de dosificación sólida curada a una temperatura de 35°C a 65°C durante un tiempo suficiente tal que se alcanza un punto final al que dicha forma de dosificación sólida proporciona un perfil de disolución estable, determinándose dicho punto final comparando el perfil de disolución de dicha forma de dosificación sólida inmediatamente después de curado con el perfil de disolución de dicha forma de dosificación sólida después de exposición a condiciones de almacenamiento acelerado de al menos un mes a 40°C y el 75% de humedad relativa.
NUEVA FORMULACION DE LIBERACION MODIFICADA.
(16/11/2006). Solicitante/s: ASTRAZENECA AB. Inventor/es: JUPPO, ANNE.
Una formulación en forma de dispersión sólida de liberación modificada, de múltiples partículas, que comprende (i) una sustancia farmacéutica activa que tiene una solubilidad en agua de, o por debajo de, 8 mg/ml a temperatura ambiente; (ii) al menos un formador de matriz hidrófobo que es un lípido anfifílico que se puede fundir, que no se hincha, que tiene una solubilidad en agua por debajo de 1 mg/g; y (iii) al menos un formador de matriz hidrófilo que es un excipiente que se puede fundir, que tiene una solubilidad en agua por encima de 0, 1 g/g; en la que la relación en peso de formador de matriz hidrófobo/formador de matriz hidrófilo es 1; y el tamaño de partículas es menor que 300 m.
COMPOSICIONES DIURETICAS DE LIBERACION PROLONGADA.
(16/11/2006). Ver ilustración. Solicitante/s: FERRER INTERNACIONAL, S.A.. Inventor/es: ROMERO VIDAL,ALFONSO, GUERRERO MARTINEZ,MARTA, GUGLIETTA,ANTONIO.
Composiciones diuréticas de liberación prolongada, que comprenden el principio activo Torasemida, las cuales suponen una gran ventaja al evitar las molestias urgencias urinarias causadas por las composiciones convencionales de liberación inmediata.
PREPARACION FARMACEUTICA PARA LA LIBERACION SOSTENIDA DE UN PRINCIPIO FARMACEUTICAMENTE ACTIVO.
(16/11/2006). Ver ilustración. Solicitante/s: LACER S.A.. Inventor/es: JURADO SANCHEZ,FRANCISCO, DE PABLO SEDANO,MARTA, ARGILAGA CAMPANO,MONICA.
Preparación farmacéutica para liberación sostenida de un(unos) principio(s) farmacéuticamente activo (s), en donde la preparación está formada por partículas con núcleo interno y un primer recubrimiento sobre el mismo, en donde dicho recubrimiento contiene una mezcla de: (a) entre el 50 % y el 95 % en peso de un copolímero de etil acrilato, metilmetacrilato y cloruro de trimetilaminoetil metacrilato en una relación molar entre los tres acrilatos de 1 : 1,8-2,2 : 0,08-0,12, preferiblemente de 1:2:0,1, (b) entre el 2 % y el 30 % en peso de un copolímero de etilacrilato y ácido metacrílico en una relación molar de 1: 0,8-1,2, preferiblemente de 1:1, y (c) entre el 1 % y el 40 % en peso del principio farmacéuticamente activo.
SUMINISTRO CONTROLADO DE ANTIDEPRESIVOS.
(01/11/2006) Forma de dosificación que comprende una pared que delimita un compartimiento , presentando la pared un orificio de salida formado o formable en la misma y siendo semipermeable por lo menos una parte de la pared ; una capa expandible dispuesta dentro del compartimiento en posición alejada del orificio de salida y en comunicación de fluidos con la parte semipermeable de la pared; una capa de fármaco dispuesta dentro del compartimiento adyacente al orificio de salida, en la que la capa de fármaco se encuentra en estado seco o sustancialmente seco para una liberación en un estado seco o sustancialmente seco, y que comprende un compuesto con la fórmula estructural siguiente: o una sal de adición de ácido farmacéuticamente aceptable del mismo, en la que R es un halógeno; y una pared secundaria entre…
COMPOSICION FARMACEUTICA QUE COMPRENDE DOXAZOSINA.
(16/10/2006). Solicitante/s: PHARMA PASS II LLC. Inventor/es: SETH, PAWAN.
Composición de doxazosina, que comprende: (a) un núcleo de tableta que comprende entre el 1% y el 5% de doxazosina y entre el 50% y el 80% en peso de óxido de polietileno que forma la matriz hidrofílica, obteniéndose dicho núcleo mediante compresión de gránulos; y opcionalmente (b) un recubrimiento.
MATRICES FORMADAS DE POLIMERO Y COMPUESTOS HIDROFOBOS PARA USO EN LA ADMINISTRACION DE FARMACOS.
(01/10/2006). Solicitante/s: ACUSPHERE, INC.. Inventor/es: BERNSTEIN, HOWARD, CHICKERING, DONALD, KHATTAK, SARWAT, STRAUB, JULIE.
Una matriz polimérica porosa para administrar un agente terapéutico o profiláctico, en la que la matriz se forma a partir de un polímero biocompatible que ha incorporado en su interior: un agente terapéutico o profiláctico una cantidad efectiva de un compuesto hidrófobo en el interior de la matriz para modificar la difusión de agua al interior de la matriz y la administración del agente terapéutico o profiláctico desde la matriz, en el que la matriz se obtiene emulsificando un agente formador de poros con un polímero disuelto en un solvente y a continuación eliminando el agente formador de poros y el solvente.
COMPOSICION FARMACEUTICA PARA LA LIBERACION CONTROLADA DE UN ANTIBIOTICO BETA-LACTAMA.
(16/09/2006). Solicitante/s: LUPIN LIMITED. Inventor/es: SEN, HIMADRI, KSHIRSAGAR, RAJESH SURESH, BHAMARE, SHAILESH SURESH.
Una composición farmacéutica estable mejorada para la liberación controlada de un ingrediente activo que comprende una matriz de liberación controlada que comprende: a) un antibiótico beta-lactama o sus hidratos, sales o ésteres farmacéuticamente aceptables como ingrediente activo en cantidades del 30% al 90% en peso; y b) una mezcla de polímeros hidrófilos en una cantidad de 1% a 25% seleccionados del grupos consistente en al menos un alginato sódico en cantidades de 0,1% a 20%, y al menos una goma xantano en cantidades de 0,1% a 20%, caracterizado porque la matriz de liberación controlada también comprende: c) una sal de calcio en una cantidad de 8% a 20% en peso de alginato sódico.
COMPOSICIONES DE ADMINISTRACION LIQUIDA DE FARMACOS.
(01/09/2006). Ver ilustración. Solicitante/s: ATRIX LABORATORIES, INC.. Inventor/es: DUNN, RICHARD L., YEWEY, GERALD L., KRINICK, NANCY L., RADOMSKY, MICHAEL L., BROUWER, GERBRAND, TIPTON, ARTHUR J..
Una composición farmacéutica de administración líquida adecuada para la formación de un implante de liberación controlada, y comprende cantidades efectivas de: a) un polímero temo plástico biodegradable biocompatible, que es insoluble en un medio acuoso; b) un solvente orgánico biocompatible para el polímero que es miscible en un medio acuoso; y c) un componente de liberación controlada que comprende un agente activo donde por contacto con el medio acuoso, la composición forma un implante gelatinoso por disipación o dispersión del solvente orgánico dentro del medio acuoso, con el componente de liberación controlada embebido en el mismo.
SISTEMA OSMOTICO DE ADMINISTRACION DE DOSIS EN FORMA SOLUBLE.
(16/06/2006). Solicitante/s: SHIRE LABORATORIES INC. Inventor/es: RUDNIC, EDWARD M., BURNSIDE, BETH A., FLANNER, HENRY, H., WASSINK, SANDRA, E., COUCH, RICHARD, A..
SE PRESENTA UN SISTEMA OSMOTICO DE ADMINISTRACION DE UNA SUBSTANCIA FARMACEUTICA QUE COMPRENDE: A) UNA PARED SEMIPERMEABLE QUE CONTIENE SU INTEGRIDAD DURANTE LA ADMINISTRACION DE LA SUBSTANCIA FARMACEUTICA Y QUE TIENE AL MENOS UN CONDUCTO A TRAVES DE LA MISMA; B) UNA COMPOSICION SIMPLE, HOMOGENEA DENTRO DE LA PARED, LA COMPOSICION CONSTA ESENCIALMENTE DE I) UN AGENTE FARMACOLOGICAMENTE ACTIVO, II) AL MENOS UN AGENTE SOLUBILIZANTE NO ENGROSABLE QUE MEJORA LA SOLUBILIDAD DEL AGENTE FARMACOLOGICAMENTE ACTIVO; III) AL MENOS UN AGENTE OSMOTICO NO ENGROSABLE; Y IV) UN AGENTE EMBEBIBLE NO ENGROSABLE DISPUESTO A TRAVES DE LA COMPOSICION QUE MEJORA EL CONTACTO DEL AREA SUPERFICIAL DEL AGENTE FARMACEUTICO CON EL FLUIDO ACUOSO ENTRANTE.
SISTEMAS DE HIDROGEL HETERODISPERSOS DE LIBERACION SOSTENIDA PARA FARMACOS INSOLUBLES.
(16/06/2006). Solicitante/s: EDWARD MENDELL CO., INC.. Inventor/es: BAICHWAL, ANAND R..
Una composición de liberación prolongada, que comprende: una cantidad eficaz de un medicamento que tiene una solubilidad de menos de aproximadamente 10 g/l, para conseguir un efecto terapéutico; un excipiente de liberación prolongada que comprende un agente gelificante, un diluyente farmacéutico inerte, siendo la relación de dicho diluyente inerte a dicho agente gelificante de 1:8 a 8:1; y un material hidrófobo seleccionado del grupo que consiste en una alquilcelulosa, un material celulósico hidrófobo, un copolímero de ésteres del ácido acrílico y metacrílico, shellac, ceras, zeína, aceites vegetales hidrogenados, y mezclas de cualquiera de los anteriores, siendo incluido dicho material hidrófobo en una cantidad efectiva para retardar la hidratación de dicho agente gelificante cuando dicha forma farmacéutica se expone a un fluido ambiental.
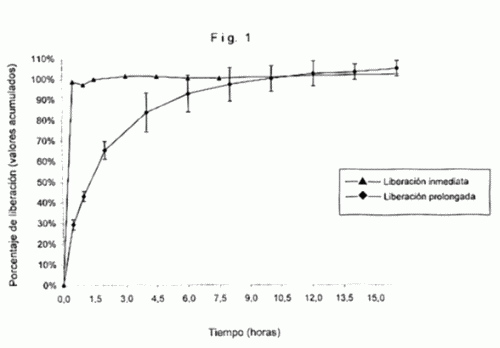
FORMULACIONES MATRICIALES PARA LIBERACION PROLONGADA DEL CLORHIDRATO DE PIRLINDOL.
(01/06/2006). Ver ilustración. Solicitante/s: TECNIMEDE-SOCIEDADE TECNICO-MEDICINAL, S.A.. Inventor/es: MENDES CERDEIRA, ANA MARIA, DE SOUSA GOUCHA JORGE, PEDRO MANUEL.
Composiciones farmacéuticas de clorhidrato de pirlindol y derivados de ácido algínico y/o derivados de una sal de ácido algínico obtenible por medio de mezcla en seco y/o granulación en seco, que permite prolongar la liberación del clorhidrato de pirlindol durante un período de tiempo seleccionado en el rango de 8 a 24 horas.
FORMA DE DOSIFICACION FARMACEUTICA ORAL DE LIBEERACION PROLONGADA.
(16/05/2006) Una forma de dosificación de liberación prolongada farmacéutica revestida entéricamente de un inhibidor de H+, K+-ATPasa, caracterizada porque la forma de dosificación comprende un material central de una matriz hidrófila o de una matriz hidrófoba que da como resultado una liberación prolongada del inhibidor de H+, K+-ATPasa durante un mínimo de 2 y un máximo de 12 horas, y el inhibidor de H+, K+-ATPasa, y porque el inhibidor de H+, K+-ATPasa es un compuesto de fórmula I, una sal alcalina del mismo, uno de los enantiómeros individuales del mismo, o una sal alcalina de uno de los enantiómeros de un compuesto de fórmula I en la que: Het1 es: Het2 es: X = en las que: N en el resto bencimidazólico significa que uno de…
ACIDO LIPOICO DE LIBERACION LENTA.
(16/04/2006). Solicitante/s: MEDICAL RESEARCH INSTITUTE. Inventor/es: BYRD, EDWARD, A., JANJIKHEL, RAJIV.
Una formulación de dosificación oral consistente en una cantidad terapéuticamente efectiva de ácido lipoico y un material excipiente, donde la formulación: (a) protege al ácido lipoico de la degradación química en el tracto gastrointestinal de un paciente y (b) libera el ácido lipoico de un modo controlado, de tal forma que el ácido lipoico se mantiene en el plasma sanguíneo en un nivel terapéutico a lo largo de un período de 4 horas o más.
PREPARADOS FARMACEUTICOS CON UNA COMBINACION DE EN CADA CASO AL MENOS UN REPRESENTANTE DE LOS ANTIBIOTICOS Y DE LOS ANTIFLOGISTICOS, PROCEDIMIENTO PARA SU PRODUCCION, ASI COMO SU USO.
(16/04/2006). Solicitante/s: HERAEUS KULZER GMBH & CO.KG. Inventor/es: VOGT, SEBASTIAN, DR., SCHNABELRAUCH, MATTHIAS, DR., KUHN, KLAUS-DIETER, DR..
Preparado farmacéutico, caracterizado porque contiene sales cuyo componente catiónico es al menos un representante de los antibióticos gentamicina, clindamicina, neomicina, estreptomicina, tetraciclina, doxiciclina, oxitetraciclina y rolitetraciclina, y cuyo componente aniónico es al menos un representante de los antiflogísticos ibuprofeno, naproxeno, indometacina, 21- fosfato de dexametasona, 21-sulfato de dexametasona, 21- sulfato de triamcinolona y 21-sulfato de triamcinolo.
VALVULA PARA DISPOSITIVOS OSMOTICOS.
(16/03/2006). Solicitante/s: ALZA CORPORATION. Inventor/es: CARR, JOHN, P., ECKENHOFF, JAMES, B..
Dispositivo osmótico de dispensación para la dispensación de un agente beneficioso, comprendiendo el dispositivo: una cápsula implantable que presenta un primer extremo y un segundo extremo , conteniendo la cápsula un agente osmótico y un agente beneficioso ; un orificio de dispensación de agente beneficioso; y un elemento de válvula ; caracterizado porque el elemento de válvula está situado en el interior de la cápsula para abrir y cerrar el orificio de dispensación de agente beneficioso mediante su movimiento a lo largo de una dirección longitudinal en el interior de la cápsula desde una posición cerrada, en la que el elemento de válvula bloquea la dispensación de agente beneficioso a través del orificio de dispensación de agente beneficioso, a una posición abierta, en la que se permite la dispensación del agente beneficioso a través del orificio de dispensación.
MEDICAMENTO QUE CONTIENE 3-(3-DIMETILAMINO-1-ETIL-2-METILPROPIL)FENOL Y QUE PROPORCIONA UNA LIBERACION RETARDADA DEL PRINCIPIO ACTIVO.
(16/03/2006) Formulación farmacéutica de liberación retardada que contiene 3-(3-dimetilamino-1-etil-2-metilpropil)fenol o una de sus sales farmacéuticamente aceptables en una matriz de liberación retardada del principio activo, donde la matriz contiene del 1 al 80% en peso de uno o varios polímeros hidrófilos o hidrófobos como formador de matriz farmacéuticamente aceptable y tiene in vitro la siguiente velocidad de liberación, medida mediante el método de Ph. Eur. Paddle a 75 r.p.m. en un tampón (según Ph. Eur.) con un pH de 6, 8 a 37ºC y con detección espectrométrica UV: 3-35% en peso (referido al 100% en peso del principio activo) de 3-(3-dimetilamino-1-etil-2-metilpropil) fenol liberado después de 0, 5 horas, 5-50% en peso de 3-(3-dimetilamino-1-etil-2-metilpropil)fenol liberado después de 1 hora, 10-75% en peso de 3-(3-dimetilamino-1-etil-2-metilpropil)fenol…
MEDICAMENTO A BASE DE TRAMADOL.
(01/03/2006). Solicitante/s: GRUNENTHAL GMBH. Inventor/es: FRIDERICHS, ELMAR, BARTHOLOMIUS, JOHANNES.
Medicamento que contiene racemato de tramadol en forma retardada y el enantiómero (+) de tramadol en forma no retardada.
FORMULACIONES FARMACEUTICAS PARA LA LIBERACION CONTROLADA DE 4-AMINO-6,7-DIMETOXI-2-(5-METANOSULFONAMIDO-1 ,2,3,4-TETRAHIDROISOQUINOL-2-IL)-5-(2-PIRIDIL)QUINAZOLINA.
(01/02/2006). Ver ilustración. Solicitante/s: PFIZER LIMITED. Inventor/es: DAVIS, JOHN, D., PFIZER GLOBAL RESEARCH AND DEV., HUMPHREY, MICHAEL, J., PFIZER GLOBAL RCH. AND DEV., MACRAE, ROSS, J., PFIZER GLOBAL RESEARCH AND DEV., SMITH, JANET, S., PFIZER GLOBAL RESEARCH AND DEV.
Una formulación farmacéutica de liberación controlada para administración oral que comprende 4-amino-6, 7-dimetoxi- 2-(5-metanosulfonamido-1 , 2, 3, 4-tetrahidroisoquinol-2-il)-5- (2-piridil)quinazolina , o una sal farmacéuticamente aceptable de la misma; y un adyuvante, diluyente o vehículo farmacéuticamente aceptable; caracterizada porque la formulación está adaptada para liberar al menos un 50% en peso de la 4-amino-6, 7-dimetoxi-2-(5-metanosulfonamido- 1, 2, 3, 4-tetrahidro-isoquinol-2-il)-5-(2- piridil)quinazolina, o la sal farmacéuticamente aceptable de la misma, después de 6 horas y menos del 30% en peso después de 1 hora en el Aparato 1 descrito en la Farmacopea de Estados Unidos 24 , páginas 1941-1943, que tiene recipientes de 1 litro, cestas de malla 40 (orificios de 0, 4 mm), una velocidad de rotación de 100 rpm y un medio de disolución que consta de 900 ml de ácido clorhídrico 0, 01 M que contiene cloruro sódico al 0, 7% p/v a 37ºC.
COMPRIMIDO MATRICIAL QUE PERMITE LA LIBERACION PROLONGADA DE TRIMETAZIDINA TRAS SU ADMINISTRACION ORAL.
(16/10/2005). Solicitante/s: LES LABORATOIRES SERVIER. Inventor/es: WUTHRICH, PATRICK, HUET DE BAROCHEZ, BRUNO, DAUPHANT, CLAUDE.
Comprimido matricial para la liberación prolongada de trimetazidina, o de una de sus sales farmacéuticamente aceptables, caracterizado porque la liberación prolongada se controla mediante un polímero derivado de celulosa presente en la matriz seleccionado entre hidroxipropilcelulosa , hidroxietilcelulosa, hidroximetilcelulosa , metilcelulosa e hidroxipropilmetilcelulosa.
PROCEDIMIENTO Y SISTEMA PARA LA LIBERACION UNIFORME DE UN FARMACO.
(16/10/2005). Ver ilustración. Solicitante/s: WARNER-LAMBERT COMPANY. Inventor/es: FESSEHAIE, MEBRAHTU, GHEBRE-SELLASSIE, ISAAC, MOLLAN, MATTHEW JOSEPH JR., MAYASSI, MONZER MICHAEL, WOLDEGABER, HAIMANOT, DYAR, STEPHEN CRAIG.
Una forma de dosificación farmacéutica que comprende un núcleo central que incluye un agente farmacéutico en una composición de liberación controlada, teniendo el mencionado núcleo central dos superficies finales opuestas expuestas, y una superficie periférica en un borde externo del mencionado núcleo que se extiende entre las dos mencionadas superficies finales opuestas, el mencionado borde periférico está rodeado por un forro limitante de la difusión, siendo el mencionado forro esencialmente impermeable al agua o a los fluidos corporales, en el que el mencionado forro limita la difusión de los fluidos hacia el interior del mencionado núcleo central, en la que la mencionada forma de dosificación farmacéutica se forma por coextrusión del mencionado núcleo central y el mencionado forro dando como resultado el mencionado núcleo central compuesto de una matriz cristalina.
COMPOSICION FARMACEUTICA CON VELOCIDAD DE LIBERACION DE FARMACO CONTROLADA.
(16/09/2005). Solicitante/s: SSP CO., LTD. Inventor/es: OKADA, MINORU, SUZUKI, MAKOTO, ISHIGAKI, KENJI, ONO, KENJI, KASAI SHUICHI, IMAMORI,KATSUMI.
ESTA INVENCION SE RELACIONA CON UNA COMPOSICION FARMACEUTICA CON UN INDICE DE LIBERACION DEL FARMACO CONTROLADO. LA COMPOSICION CONSTA DE UNA MATRIZ FORMADA POR LOS SIGUIENTES INGREDIENTES: (A) UNA SUSTANCIA BIODEGRADABLE, BIOCOMPATIBLE, ALTAMENTE MOLECULAR Y/O IONES METALICOS POLIVALENTES O UNA FUENTE DE IONES METALICOS POLIVALENTES Y; (B) ACIDO HIALURICO O UNA SAL DEL MISMO Y UN FARMACO INCORPORADO COMO INGREDIENTE (C) A LA CITADA MATRIZ. LA COMPOSICION ES BIODEGRADABLE Y BIOCOMPATIBLE Y PERMITE UN FACIL CONTROL DEL INDICE DE LIBERACION DEL FARMACO Y PUEDE MOSTRAR PERSISTENTEMENTE SU EFECTO FARMACOLOGICO DURANTE UN PERIODO DE TIEMPO PROLONGADO.
PREPARACION DE LEUPRORELINA DE LIBERACION SOSTENIDA.
(01/09/2005). Solicitante/s: TAKEDA CHEMICAL INDUSTRIES, LTD.. Inventor/es: KAMEI, SHIGERU, IGARI, YASUTAKA, OGAWA, YASUAKI.
Un método para producir microcápsulas que comprenden una preparación de liberación sostenida de leuprorelina o una sal de la misma, que comprende (a) disolver o suspender leuprorelina o una sal de la misma en una solución en disolvente orgánico de un polímero biodegradable que comprende un copolímero de ácido láctico y ácido glicólico que tiene un grupo carboxilo terminal; (b) añadir la mezcla a un medio acuoso para proporcionar una emulsión Ac./Ag.; y (c) transformar la emulsión en microcápsulas por separación del disolvente orgánico.
PROCEDIMIENTO PARA LA OBTENCION DE FORMAS DE ADMINISTRACION SOLIDAS ORALES CON LIBERACION DE PRODUCTO ACTIVO RETARDADA.
(16/07/2005). Solicitante/s: BASF AKTIENGESELLSCHAFT. Inventor/es: KOLTER, KARL, DR., FLICK, DIETER, ASCHERL, HERMANN.
Procedimiento para la obtención de una forma de administración oral o liberación de producto activo retardada, que contiene a) una mezcla formulada de acetato de polivinilo y polivinilpirrolidona , b) al menos un producto activo, c) en caso dado polímeros hidrosolubles o aditivos lipófilos de peso molecular reducido o elevado, d) así como, en caso dado, otras substancias auxiliares habituales, caracterizado porque la proporción de acetato de polivinilo respecto a polivinilpirrolidona asciende a 6 : 4 hasta 9 : 1, acetato de polivinilo y polivinilpirrolidona presentan respectivamente un peso molecular de 20.000 a 1.000.000, y el granulado de la mezcla de a) a d) o a) a c) o a) y b) y d) o a) y b) se efectúa sin adición de disolvente mediante calentamiento a 40ºC hasta 130ºC.
COMPOSICION FARMACEUTICA QUE COMPRENDE UN AGONISTA DEL RECEPTOR 5HT1.
(16/07/2005) Una composición farmacéutica de una forma farmacéutica sólida recubierta para administración por vía oral y que en la administración se disgrega y se dispersa dentro del tracto gastrointestinal, que comprende sumatriptán o una de sus sales o solvatos farmacéuticamente aceptable como ingrediente activo, en la que la cantidad de sumatriptán, expresada como el peso de la base libre, está en el intervalo de 20 mg a 150 mg; y que comprende el componente básico de un par efervescente, un disgregante y un material de carga insoluble, en la que el componente básico comprende de 5 a 50% en peso, el disgregante comprende de 0, 5 a 10% en peso y el material de carga insoluble comprende de 35…
(16/07/2005) Forma de dosificación oral de liberación sostenida que comprende: (a) una pared que define un compartimiento, comprendiendo la pared una capa semipermeable; (b) una capa expandible dispuesta en el compartimiento y en comunicación fluida con la capa semipermeable; (c) una cápsula dispuesta en el compartimiento y en relación de contacto directo o indirecto con la capa expandible, comprendiendo la cápsula una composición líquida de un fármaco antiviral que comprende un fármaco antiviral solubilizado en un solvente, en la que dicho fármaco antiviral está presente en una cantidad del 5% en peso al 60% en peso y el solvente está presente en una cantidad del 20% en peso al 95% en peso de la composición total del fármaco antiviral, estando dicha composición sustancialmente…
COMPOSICIONES DE LIBERACION CONTROLADA PARA EL TRATAMIENTO DE HIPERLIPIDEMIA.
(01/07/2005). Solicitante/s: FUISZ INTERNATIONAL LTD. Inventor/es: MYERS, MICHAEL, SANGHVI, PRADEEPKUMAR, P.
Unidades de dosificación baja para utilizarse una vez al día que producen efectos secundarios mínimos. Contienen una combinación de microesferas formuladas para liberar conjuntamente un componente inhibidor de HMG-CoA reductasa y un componente de niacina.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}