Métodos para determinar variantes de secuencias usando secuenciación ultraprofunda.
Un método para la detección de variantes de secuencia que tienen una frecuencia de menos de 5% en una población de ácidos nucleicos,
comprendiendo el método las etapas de:
(a) amplificar un segmento de polinucleótido común a dicha población de ácido nucleico con un par de cebadores de ácidos nucleicos para PCR que definen un locus para producir una primera población de amplicones comprendiendo cada amplicón dicho segmento de polinucleótido;
(b) liberar la primera población de amplicones en microrreactores acuosos en una emulsión de agua-en-aceite de manera que una pluralidad de los microrreactores acuosos comprende (1) un único amplicón de la primera población de amplicones, (2) una única perla, y (3) disolución de reacción de amplificación que contiene los reactivos necesarios para realizar la amplificación de ácido nucleico;
(c) amplificar clonalmente cada miembro de dicha primera población de amplicones mediante reacción en cadena de la polimerasa para producir una pluralidad de poblaciones de segundos amplicones en donde cada población de segundos amplicones deriva de un miembro de dicha primera población de amplicones;
(d) inmovilizar dichos segundos amplicones en una pluralidad de las perlas en los microrreactores de manera que cada perla comprenda una población de dichos segundos amplicones;
(e) romper la emulsión para recuperar las perlas de los microrreactores;
(f) determinar en paralelo una secuencia de ácido nucleico para los segundos amplicones en cada perla, a una profundidad (es decir, número de lecturas de secuencia individuales) de más de 100 para producir una población de secuencias de ácido nucleico; y
(g) determinar una incidencia de cada tipo de nucleótido en cada posición de dicho segmento de polinucleótido para detectar dichas variantes de secuencia en dicha población de ácido nucleico;
en donde el método no requiere conocimiento previo de la composición de secuencia de ácido nucleico de las variantes de secuencia,
y en donde dichos cebadores de ácido nucleico son cebadores bipartitos que comprenden una región 5' y una región 3', en donde dicha región 3' es complementaria a una región en dicho segmento de polinucleótido y en donde dicha región 5' es homóloga a un cebador de secuenciación o un complemento del mismo.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/US2006/013753.
Solicitante: 454 LIFE SCIENCES CORPORATION.
Nacionalidad solicitante: Estados Unidos de América.
Dirección: 20 COMMERCIAL STREET BRANFORD CT 06405 ESTADOS UNIDOS DE AMERICA.
Inventor/es: EGHOLM, MICHAEL, LOHMAN,KENTON, ROTHBERG,JONATHAN, LEAMON,JOHN HARRIS, LEE,WILLIAM LUN, SIMONS,JAN FREDRICK, DESANY,BRIAN, RONAN,MIKE TODD, DRAKE,JAMES.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C12Q1/68 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12Q PROCESOS DE MEDIDA, INVESTIGACION O ANALISIS EN LOS QUE INTERVIENEN ENZIMAS, ÁCIDOS NUCLEICOS O MICROORGANISMOS (ensayos inmunológicos G01N 33/53 ); COMPOSICIONES O PAPELES REACTIVOS PARA ESTE FIN; PROCESOS PARA PREPARAR ESTAS COMPOSICIONES; PROCESOS DE CONTROL SENSIBLES A LAS CONDICIONES DEL MEDIO EN LOS PROCESOS MICROBIOLOGICOS O ENZIMOLOGICOS. › C12Q 1/00 Procesos de medida, investigación o análisis en los que intervienen enzimas, ácidos nucleicos o microorganismos (aparatos de medida, investigación o análisis con medios de medida o detección de las condiciones del medio, p. ej. contadores de colonias, C12M 1/34 ); Composiciones para este fin; Procesos para preparar estas composiciones. › en los que intervienen ácidos nucleicos.
PDF original: ES-2404311_T3.pdf
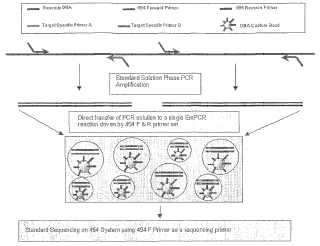
Fragmento de la descripción:
Métodos para determinar variantes de secuencias usando secuenciación ultraprofunda.
Campo de la invención La invención proporciona un método para detectar y analizar variantes de secuencias de baja frecuencia, incluyendo polimorfismos de nucleótido único (SNP) , variantes de inserción/deleción (referidos como "indeles") y frecuencias alélicas, en una población de polinucleótidos diana en paralelo.
Antecedentes de la invención El ADN genómico varía significativamente de un individuo a otro, excepto en hermanos idénticos. Muchas enfermedades humanas surgen de las variaciones genómicas. La diversidad genética entre los seres humanos y otras formas de vida explica las variaciones hereditarias observadas en la susceptibilidad a enfermedades. Las enfermedades derivadas de estas variaciones genéticas son la enfermedad de Huntington, la fibrosis quística, la distrofia muscular de Duchenne, y ciertas formas de cáncer de mama. Cada una de estas enfermedades está asociada con una mutación de un único gen. Enfermedades tales como la esclerosis múltiple, la diabetes, el Parkinson, la enfermedad de Alzheimer, y la hipertensión son mucho más complejas. Estas enfermedades pueden estar debidas a causas poligénicas (influencias de múltiples genes) o multifactoriales (influencias de múltiples genes y ambientales) . Muchas de las variaciones en el genoma no dan como resultado un rasgo de una enfermedad. Sin embargo, como se ha descrito anteriormente, una sola mutación puede dar lugar a un rasgo de una enfermedad. La capacidad de explorar el genoma humano para identificar la ubicación de los genes que subyacen o están asociados con la patología de estas enfermedades es una herramienta muy poderosa en la medicina y la biología humana.
Varios tipos de variaciones de la secuencia, incluyendo inserciones y deleciones (indeles) , diferencias en el número de secuencias repetidas, y diferencias de pares de bases individuales (SNP) dan como resultado la diversidad genómica. Las diferencias de pares de bases individuales, referidas como polimorfismos de nucleótido único (SNP) son el tipo más frecuente de variación en el genoma humano (presentándose en aproximadamente 1 de cada 103 bases) . Según se utiliza en la presente memoria, un SNP puede ser cualquier posición genómica en la cual aparecen al menos dos o más alelos de nucleótidos alternativos. Según se utiliza la presente memoria, un SNP puede también referirse a cualquier variante inserción/deleción de base única (referida como "indel") , o un indel que incluye la inserción y/o deleción de entre 2 y 100 o más bases. Los SNP son muy adecuados para el estudio de variaciones de la secuencia, ya que son relativamente estables (es decir, exhiben tasas de mutación bajas) y debido a que pueden ser responsables de los rasgos heredados. Se entiende que en la discusión anterior, el término SNP también está destinado a ser aplicable a "indel" (definido a continuación) .
Los polimorfismos identificados utilizando análisis basado en microsatélites, por ejemplo, se han utilizado para una variedad de propósitos. El uso de estrategias de ligamiento genético para identificar las ubicaciones de los factores mendelianos individuales ha tenido éxito en muchos casos (Benomar et al. (1995) , Nat. Genet., 10:84-8; Blanton et al. (1991) , Genomics, 11:857-69) . La identificación de las localizaciones cromosómicas de los genes supresores de tumores en general se ha logrado mediante el estudio de la pérdida de heterozigosidad en tumores humanos (Cavenee et al. (1983) , Nature, 305:779-784; Collins et al. (1996) , Proc. Natl. Acad. Sci. USA, 93:14771-14775; Koufos et al. (1984) , Nature, 309:170-172; y Legius et al. (1993) , Nat. Genet., 3:122-126) . Además, el uso de marcadores genéticos para inferir las posiciones cromosómicas de los genes que contribuyen a rasgos complejos, tales como diabetes de tipo I (Davis et al. (1994) , Nature, 371:130-136; Todd et al. (1995) , Proc. Natl. Acad. Sci. EE.UU., 92:8560-8565) , se ha convertido en un foco de investigación en genética humana.
Si bien se ha avanzado considerablemente en la identificación de las bases genéticas de muchas enfermedades humanas, las metodologías actuales utilizadas para desarrollar esta información están limitadas por los costes prohibitivos y la gran cantidad de trabajo necesario para obtener información genotípica de grandes poblaciones de muestras. Estas limitaciones hacen que la identificación de mutaciones genéticas complejas que contribuyen a trastornos tales como la diabetes sea extremadamente difícil. Las técnicas para explorar el genoma humano para identificar las ubicaciones de los genes implicados en los procesos de enfermedad comenzaron a principios de la década de 1980 con el uso de polimorfismo de longitud de fragmentos de restricción (RFLP) , (Botstein et al. (1980) , Am. J. Hum. Genet., 32:314-31; Nakamura et al. (1987) , Science, 235:1616-22) . El análisis RFLP consiste en técnicas de transferencia Southern y otras. La transferencia de Southern es cara y lleva mucho tiempo cuando se realiza en un gran número de muestras, tales como las requeridas para identificar un genotipo complejo asociado con un fenotipo particular. Algunos de estos problemas se evitaron con el desarrollo de análisis marcadores de microsatélites basados en la reacción en cadena de la polimerasa (PCR) . Los marcadores microsatélites son polimorfismos de longitud de secuencia simples (SSLP) que consisten en di-, tri-, y tetra-nucleótidos repetidos.
Otros tipos de análisis genómico se basan en el uso de marcadores que hibridan con las regiones hipervariables del ADN que tienen variación multialélica y alta heterozigosidad. Las regiones variables que son útiles para la huella dactilar de ADN genómico son repeticiones en tándem de una secuencia corta referida como minisatélite. El polimorfismo es debido a las diferencias alélicas en el número de repeticiones, que pueden surgir como resultado de intercambios mitóticos o meióticos desiguales o por el deslizamiento de ADN durante la replicación.
Actualmente, la identificación de las variaciones mediante secuenciación del ADN se ve obstaculizada por una serie de deficiencias. En los métodos actuales, la amplificación de una región de interés está seguida por secuenciación directa del producto de amplificación (es decir, una mezcla de secuencias variantes) . Alternativamente, la etapa de secuenciación está precedida por una etapa de subclonación microbiana, es decir, por inserción recombinante de productos de amplificación en un vector adecuado para la propagación en el organismo anfitrión deseado.
La desventaja de la secuenciación directa del producto de amplificación consiste en una señal mixta que se produce en sitios variables en la secuencia. Las contribuciones relativas de los diferentes nucleótidos en dichas señales mixtas son difíciles o imposibles de cuantificar, incluso cuando la frecuencia del alelo de menor abundancia se aproxima a 50%. Además, si la variación es una inserción o deleción (en lugar de una sustitución de bases) , el desplazamiento de fase resultante entre las diferentes moléculas dará lugar a una señal desordenada, ilegible.
La adición de una etapa de clonación microbiana supera los problemas asociados con la secuenciación directa, ya que no se encuentran señales mixtas. Sin embargo, esta estrategia requiere un mayor número de reacciones de secuenciación. Además, la etapa de clonación microbiana es costosa y consume tiempo, y puede también evitar ciertas variantes, y por tanto distorsionar la frecuencia relativa de las variantes. Si se desea la secuenciación de un gran número (es decir, cientos, miles, decenas de miles) de clones, el coste es extremadamente alto.
Cada uno de estos métodos actuales tiene inconvenientes importantes, ya que consumen mucho tiempo y tienen una resolución limitada. Si bien la secuenciación del ADN proporciona la mayor resolución, también es el método más costoso para la determinación de SNP. En este momento, la determinación de la frecuencia de SNP entre una población de 1.000 diferentes muestras es muy costosa y la determinación de la frecuencia de SNP entre una población de 100.000 muestras es prohibitiva. Por lo tanto, existe una necesidad continua en la técnica de métodos económicos de identificación y resecuenciación de variantes de secuencia presentes en las poblaciones de polinucleótidos, especialmente variantes presentes a frecuencias bajas.
Da Mota et al. (2002) , Eur. J. Immunogen, 29:223-227 describen dos enfoques experimentales de genotipificación para identificar alelos del gen BoLA-DRB3 de ganado cebú de raza lechera tropical: (i) secuenciación directa de productos génicos de PCR, y (ii) secuenciación... [Seguir leyendo]
Reivindicaciones:
1. Un método para la detección de variantes de secuencia que tienen una frecuencia de menos de 5% en una población de ácidos nucleicos, comprendiendo el método las etapas de:
(a) amplificar un segmento de polinucleótido común a dicha población de ácido nucleico con un par de cebadores de ácidos nucleicos para PCR que definen un locus para producir una primera población de amplicones comprendiendo cada amplicón dicho segmento de polinucleótido;
(b) liberar la primera población de amplicones en microrreactores acuosos en una emulsión de agua-enaceite de manera que una pluralidad de los microrreactores acuosos comprende (1) un único amplicón de la primera población de amplicones, (2) una única perla, y (3) disolución de reacción de amplificación que contiene los reactivos necesarios para realizar la amplificación de ácido nucleico;
(c) amplificar clonalmente cada miembro de dicha primera población de amplicones mediante reacción en cadena de la polimerasa para producir una pluralidad de poblaciones de segundos amplicones en donde cada población de segundos amplicones deriva de un miembro de dicha primera población de amplicones;
(d) inmovilizar dichos segundos amplicones en una pluralidad de las perlas en los microrreactores de manera que cada perla comprenda una población de dichos segundos amplicones;
(e) romper la emulsión para recuperar las perlas de los microrreactores;
(f) determinar en paralelo una secuencia de ácido nucleico para los segundos amplicones en cada perla, a una profundidad (es decir, número de lecturas de secuencia individuales) de más de 100 para producir una población de secuencias de ácido nucleico; y
(g) determinar una incidencia de cada tipo de nucleótido en cada posición de dicho segmento de polinucleótido para detectar dichas variantes de secuencia en dicha población de ácido nucleico;
en donde el método no requiere conocimiento previo de la composición de secuencia de ácido nucleico de las variantes de secuencia,
y en donde dichos cebadores de ácido nucleico son cebadores bipartitos que comprenden una región 5' y una región 3', en donde dicha región 3' es complementaria a una región en dicho segmento de polinucleótido y en donde dicha región 5' es homóloga a un cebador de secuenciación o un complemento del mismo.
2. Un método de acuerdo con la reivindicación 1, en donde:
la etapa (e) comprende la liberación de las perlas con los segundos amplicones unidos a una formación de cámaras de reacción sobre una superficie plana y la realización de una reacción de secuenciación de forma simultánea sobre la pluralidad de cámaras de reacción para determinar una pluralidad de secuencias de ácido nucleico correspondientes a una pluralidad de alelos.
3. Un método de acuerdo con la reivindicación 1 o 2, en donde el método comprende determinar la secuencia de nucleótidos de una región de polinucleótidos de interés a una profundidad (es decir, número de lecturas de secuencias individuales) de más de 1000.
4. El método de la reivindicación 1, en donde dicha región 5' es homóloga a un oligonucleótido de captura o un complemento del mismo sobre dicho soporte sólido móvil.
5. El método de la reivindicación 1, en donde dicha perla tiene un diámetro seleccionado del grupo que consiste en aproximadamente 1 a aproximadamente 500 micras, entre aproximadamente 5 y aproximadamente 100 micras, entre aproximadamente 10 y aproximadamente 30 micras y entre aproximadamente 15 y aproximadamente 25 micras.
6. El método de la reivindicación 1, en donde dicha perla comprende un oligonucleótido que hibrida e inmoviliza dicha primera población de amplicones, segundos amplicones, o ambos.
7. El método de la reivindicación 1, en donde dicha etapa de determinación de una secuencia de ácido nucleico se lleva a cabo liberando la pluralidad de soportes sólidos móviles en una formación de al menos 10.000 cámaras de reacción sobre una superficie plana, en donde una pluralidad de las cámaras de reacción comprenden no más de un único soporte sólido móvil; y determinando una secuencia de ácido nucleico de los amplicones sobre cada uno de dichos soportes sólidos móviles.
8. El método de la reivindicación 1, en donde dicha etapa de determinación de una secuencia de ácido nucleico se lleva a cabo realiza mediante secuenciación basada en pirofosfato.
9. El método de la reivindicación 1, en donde dicha población de ácido nucleico comprende ADN, ARN, ADNc o una combinación de los mismos.
10. El método de la reivindicación 1, en donde la población de ácido nucleico deriva de una pluralidad de organismos.
11. El método de la reivindicación 1, en donde la población de ácido nucleico deriva de un organismo.
12. El método de la reivindicación 11, en donde dicha población de ácido nucleico deriva de múltiples muestras de tejido de dicho organismo.
13. El método de la reivindicación 11, en donde dicha población de ácido nucleico deriva de un solo tejido de dicho organismo.
14. El método de la reivindicación 1, en donde la población de ácido nucleico es de un tejido enfermo.
15. El método de la reivindicación 14, en donde dicho tejido enfermo comprende tejido tumoral.
16. El método de la reivindicación 1, en donde dicha población de ácido nucleico deriva de un cultivo bacteriano, un cultivo viral, o de una muestra ambiental.
17. El método de la reivindicación 1, en donde la primera población de amplicones tiene de 30 a 500 bases de longitud.
18. El método de la reivindicación 1, en donde dicha primera población de amplicones comprende más de 1000 amplicones, más de 5000 amplicones, o más de 10000 amplicones.
19. El método de la reivindicación 1, en donde cada uno de dichos soportes sólidos se une a al menos 10.000 miembros de dicha pluralidad de segundos amplicones.
20. El método de la reivindicación 1, en donde la secuencia de ácido nucleico de dicho segmento de polinucleótido es indeterminado o parcialmente indeterminado antes de dicho método.
21. Un método para identificar una distribución de organismos en una población que comprende una pluralidad de organismos individuales diferentes, comprendiendo el método el uso de una muestra de ácido nucleico de dicha población y que comprende las etapas de:
(a) determinar las variantes de secuencia de un segmento de ácido nucleico que comprende un locus común a todos los organismos de dicha población utilizando el método de la reivindicación 1, en donde cada organismo comprende una secuencia de ácido nucleico diferente en dicho locus; y
(b) identificar la distribución de los organismos en dicha población basándose en dicha población de secuencias de ácido nucleico.
22. El método de la reivindicación 21, en donde dicha población es una población de organismos seleccionados del grupo que consiste de bacterias, virus, organismos unicelulares, plantas y levaduras.
23. Un método para determinar una composición de una muestra de tejido, comprendiendo el método la utilización de una muestra de ácido nucleico de dicha muestra de tejido y que comprende las etapas de:
(a) detectar variantes de secuencia de un segmento de ácido nucleico utilizando el método de la reivindicación 1, en donde dicho segmento comprende un locus común a todas las células en dicha muestra de tejido y en donde cada tipo de célula comprende una variante de secuencia diferente en dicho locus; y
(b) determinar la composición de dicha muestra de tejido a partir de dicha frecuencia de nucleótidos.
Patentes similares o relacionadas:
Método para analizar ácido nucleico molde, método para analizar sustancia objetivo, kit de análisis para ácido nucleico molde o sustancia objetivo y analizador para ácido nucleico molde o sustancia objetivo, del 29 de Julio de 2020, de Kabushiki Kaisha DNAFORM: Un método para analizar un ácido nucleico molde, que comprende las etapas de: fraccionar una muestra que comprende un ácido nucleico molde […]
MÉTODOS PARA EL DIAGNÓSTICO DE ENFERMOS ATÓPICOS SENSIBLES A COMPONENTES ALERGÉNICOS DEL POLEN DE OLEA EUROPAEA (OLIVO), del 23 de Julio de 2020, de SERVICIO ANDALUZ DE SALUD: Biomarcadores y método para el diagnostico, estratificación, seguimiento y pronostico de la evolución de la enfermedad alérgica a polen del olivo, kit […]
Detección de interacciones proteína a proteína, del 15 de Julio de 2020, de THE GOVERNING COUNCIL OF THE UNIVERSITY OF TORONTO: Un método para medir cuantitativamente la fuerza y la afinidad de una interacción entre una primera proteína de membrana o parte de la misma y una […]
Secuenciación dirigida y filtrado de UID, del 15 de Julio de 2020, de F. HOFFMANN-LA ROCHE AG: Un procedimiento para generar una biblioteca de polinucleótidos que comprende: (a) generar una primera secuencia del complemento (CS) de un polinucleótido diana a partir […]
Métodos para la recopilación, estabilización y conservación de muestras, del 8 de Julio de 2020, de Drawbridge Health, Inc: Un método para estabilizar uno o más componentes biológicos de una muestra biológica de un sujeto, comprendiendo el método obtener un […]
Evento de maíz DP-004114-3 y métodos para la detección del mismo, del 1 de Julio de 2020, de PIONEER HI-BRED INTERNATIONAL, INC.: Un amplicón que consiste en la secuencia de ácido nucleico de la SEQ ID NO: 32 o el complemento de longitud completa del mismo.
Aislamiento de ácidos nucleicos, del 24 de Junio de 2020, de REVOLUGEN LIMITED: Un método de aislamiento de ácidos nucleicos que comprenden ADN de material biológico, comprendiendo el método las etapas que consisten en: (i) efectuar un lisado […]
Composiciones para modular la expresión de SOD-1, del 24 de Junio de 2020, de Biogen MA Inc: Un compuesto antisentido según la siguiente fórmula: mCes Aeo Ges Geo Aes Tds Ads mCds Ads Tds Tds Tds mCds Tds Ads mCeo Aes Geo mCes Te (secuencia […]