CIP2021: Invenciones publicadas en esta sección.
POLICETIDOS Y SU SINTESIS Y USO.
(16/04/2006). Solicitante/s: BIOTICA TECHNOLOGY LIMITED. Inventor/es: LEADLAY, PETER, FRANCIS, STAUNTON, JAMES, KHAW, LAKE, EE.
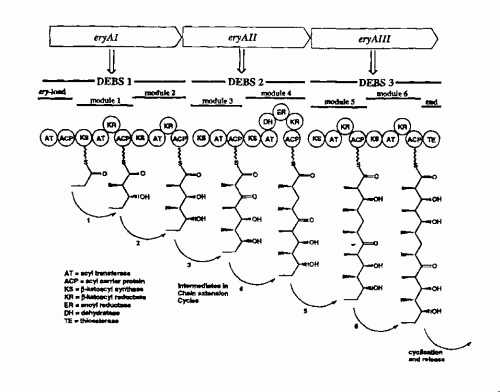
Procedimiento para producir un policétido que comprende modificar un grupo génico implicado en la biosíntesis de un policétido seleccionado entre rapamicina, FK506 e inmunomicina, incluyendo dicho grupo génico un gen ("el gen precursor") responsable de la producción de una enzima que es responsable de la producción de L-pipecolato, comprendiendo dicho procedimiento: (a) la etapa de eliminar o inactivar dicho gen precursor para producir un grupo génico modificado; y (b) expresar el grupo génico modificado en presencia de un compuesto precursor alternativo que se selecciona entre L-prolina, L-trans-4-hidroxiprolina , L-cis-4-hidroxiprolina , L-cis-3-hidroxiprolina y ácido trans-3-aza-biciclo[3 ,l,0]hexano-2-carboxílico , de manera que dicho compuesto precursor alternativo se incorpora en lugar de L-pipecolato, de modo que se produce un policétido alternativo.
PROCEDIMIENTO PARA PREPARAR UN COMPUESTO DE ERITROMICINA.
(01/04/2006). Ver ilustración. Solicitante/s: UBE INDUSTRIES, LTD. CHUGAI SEIYAKU KABUSHIKI KAISHA. Inventor/es: MIYATA, HIROYUKI, TAKAMA, AKIRA, YAMAMOTO, YASUHITO, ATAKA, KIKUO.
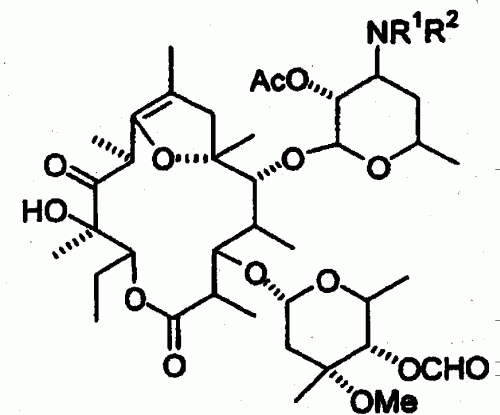
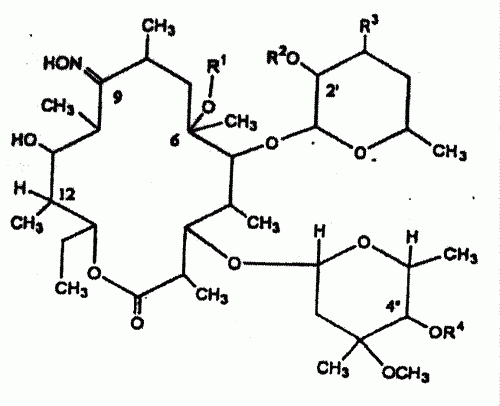
Un procedimiento para preparar un compuesto de eritromicina que comprende someter el compuesto 1 6, 9- hemicetal de 2-O-acetil-4- O-formil-11-oxo-8, 9- anhidroeritromicina A representado por la fórmula : en la que R1 y R2 cada uno de ellos representa un grupo alquilo inferior, a alquilación, en la que el compuesto 1 se hace reaccionar con un agente alquilante en presencia de una base en un disolvente mixto de 0, 18 a 1, 05 equivalentes de agua basándose en la cantidad de compuesto 1 y un disolvente orgánico, proporcionando el compuesto 6, 9- hemiacetal de 2-O- acetil-4-O-formil-11-oxo-12-alcoxi- 8, 9-anhidroeritromicina A (compuesto 3) representado por la fórmula : en la que R representa un grupo alquilo inferior, R1 y R2 tienen los mismos significados que se han definido anteriormente.
PROCEDIMIENTO DE ARILACION PARA LA FUNCIONALIZACION DE DERIVADOS DE ERITROMICINA O-ALILICA.
(16/02/2006). Solicitante/s: ABBOTT LABORATORIES. Inventor/es: ZHANG, WEIJIANG, HSU, MARGARET, CHI-PING, HAIGHT, ANTHONY, R., PETERSON, MATTHEW, JOHN, NARAYANAN, BIKSHANDARKOIL, A.
Un método para la arilación sin fosfina de un derivado de de eritromicina O-alílico, que comprende las etapas de: hacer reaccionar el grupo alilo de un derivado de O- alil-eritromicina con un agente de arilación en presencia de una base inorgánica, un catalizador de transferencia de fases y menos de un seis por ciento en moles de un catalizador de paladio en un disolvente orgánico, sin la adición de una fosfina, a una temperatura de aproximadamente 90ºC a aproximadamente 120ºC para formar un derivado de O-alquenilaril-eritromicina; y después, aislar opcionalmente dicho derivado de O- alquenilarileritromicina.
CETOLIDOS 6-O-SUSTITUIDOS QUE TIENEN ACTIVIDAD ANTIBACTERIANA.
(16/12/2005). Ver ilustración. Solicitante/s: ABBOTT LABORATORIES. Inventor/es: OR, YAT SUN, MA, ZHENKUN, CLARK, RICHARD, F., CHU, DANIEL, T., PLATTNER, JACOB, J..
La presente invención proporciona una clase novedosa de derivados de eritromicina 6-O-sustituidos que poseen una estabilidad a los ácidos incrementada en relación con la eritromicina A y la 6-O-metileritromicina A y una actividad aumentada hacia las bacterias gram negativas y las bacterias gram positivas resistentes a los macrólidos.
UN PROCEDIMIENTO PARA LA OBTENCION DE CLARITROMICINA.
(16/12/2005). Solicitante/s: ERCROS INDUSTRIAL, S.A.. Inventor/es: ASENSIO DOMINGUEZ,RAMON, CRUZADO RODRIGUEZ,MARIA CARMEN, BRUNET ROMERO,ERNESTO, RODRIGUEZ URBIS,JUAN CARLOS.
La presente invención describe la obtención de derivados de eritromicina A, que sirven de intermedios para la obtención de 6- O-metil-eritromicina A, denominada claritromicina, particularmente, a través de la obtención y el empleo de 9-O-aril derivados de la 9-oxima de la eritromicina A y la posterior purificación del producto crudo a través de la formación de una sal de tiocianato.
PROCEDIMIENTO PARA LA PREPARACION DE POLIMORFOS DE CLARITROMICINA Y NUEVO POLIMORFO IV.
(16/12/2005). Solicitante/s: TEVA PHARMACEUTICAL INDUSTRIES LTD.. Inventor/es: LIFSHITZ, IGOR, AVRUTOV, ILYA, SCHWARTZ, EDI, MASARWA, BASEM, BOROCHOVITZ, RONEN.
Procedimiento para convertir la Forma 0 polimórfica de claritromicina en la Forma II polimórfica de claritromicina, que comprende la puesta en suspensión de un sólido que comprende claritromicina de Forma 0 en agua.
DERIVADOS DE KETLIDO 2-HALO-6-0-SUSTITUIDOS.
(01/12/2005). Ver ilustración. Solicitante/s: ABBOTT LABORATORIES. Inventor/es: CHEN, YAN, OR, YAT SUN, CLARK, RICHARD, F., CHU, DANIEL, T., PHAN, LY, TAM, PLATTNER, JACOB, J..
Un compuesto que tiene la fórmula: o una sal farmacéuticamente aceptable del mismo, donde Rp es hidrógeno o un grupo hidroxi protector; X es F, Cl o Br; y R1 se selecciona entre el grupo compuesto por -CH2-CH=CH-Y, en la que Y se selecciona entre el grupo compuesto por (a) H, (b) fenilo, (c) quinolinilo, (d) naftiridinilo, (e) heteroarilo sustituido, donde el heteroarilo es piridinilo, quinolinilo o tiofenilo sustituido con carboxamida, alquilamino, dialquilamino, carboxaldehído, nitro, -CN, -CH(=N-OH), -CH(=N-NH2), -CH(=N-N=C(CH3)2), piridilo, tiofenilo o pirimidinilo o amino N-protegido, donde el grupo N-protector es t- butiloxicarbonilo; (f) quinoxalinilo, y (g) (aril)oilo, donde el arilo es fenilo; y -CH2-C=C-Y, en la que Y es como se ha definido anteriormente.
ERITROMICINAS Y PROCEDIMIENTO PARA SU PREPARACION.
(01/12/2005). Ver ilustración. Solicitante/s: BIOTICA TECHNOLOGY LIMITED PFIZER INC. Inventor/es: PACEY, MICHAEL STEPHEN, LEADLAY, PETER, FRANCIS, STAUNTON, JAMES, CORTES, JESUS.
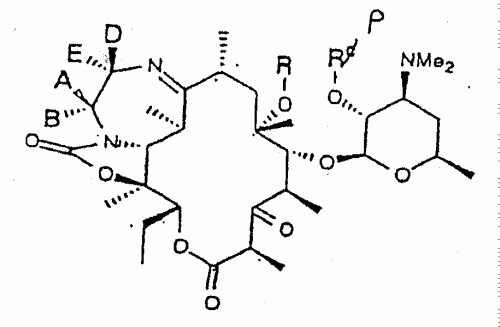
LAS ERITROMICINAS, PARTICULARMENTE CON SUSTITUYENTES R1 A LA ALTURA DEL C-13 (POR EJEMPLO, GRUPOS CICLOALQUILOS O CICLOALQUENILOS C 3 - C 6 ) SE PREPARAN HACIENDO FERMENTAR ORGANISMOS ADECUADOS EN PRESENCIA DE R 1 CO 2 H. UN ORGANISMO PREFERIDO ES LA SACCHAROPOLYSPORA ERYTHRAEA, QUE CONTENGA PREFERENTEMENTE UN PLASMIDIO INTEGRADO CAPAZ DE DIRIGIR LA SINTESIS DE LOS COMPUESTOS DESEADOS.
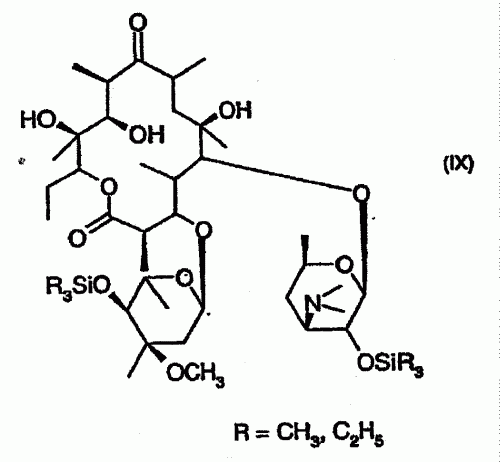
PROCEDIMIENTO PARA LA PRODUCCION DE DESCLARITROMICINA, ASI COMO PRODCUTOS INTERMEDIOS.
(01/12/2005). Ver ilustración. Solicitante/s: AVENTIS PHARMA DEUTSCHLAND GMBH. Inventor/es: JENDRALLA, HEINER, KORB, GERHARD, MUELLER-LEHAR, JURGEN.
Procedimiento para la producción de desclaritromicina, caracterizado porque a) se hace reaccionar eritromicina A con R3SiCl y/o R3Si-imidazol o o (R3Si)2NH o R3SiO3SCF3, donde R significa CH3, C2H5 bajo condiciones básicas, para obtener compuestos de la fórmula (IX), y b) R = CHA, C2H6 seguidamente se oxida mediante la adición de un agente de oxidación, obteniéndose un compuesto de la fórmula X.
CETOLIDOS 6-O-SUSTITUIDOS QUE TIENEN ACTIVIDAD ANTIBACTERIANA.
(16/11/2005) En una realización de la presente invención están los compuestos que tienen la fórmula II, donde X, Y, R, Ra y Rc se describen como antes. Un compuesto representativo de fórmula II es el Compuesto de Fórmula (II), R a es OH, R c es benzoilo, R es alilo. En una realización preferida de los compuestos de fórmula II de la invención están los compuestos en los que R a es hidroxi y R c es hidrógeno. En una realización más preferida de los compuestos de fórmula II de la invención están los compuestos que tienen la fórmula VIII, donde X es O o NOH, y R se define como antes. Entre los compuestos representativos de esta realización se incluyen, pero no están limitados a: Compuesto de Fórmula (VIII): X es O, R es alilo; Compuesto de Fórmula (VIII): X es NOH, R es alilo; Compuesto de Fórmula (VIII): X…
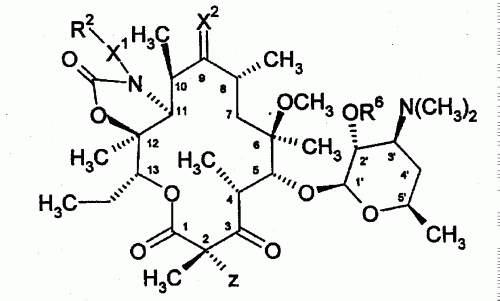
ANTIBIOTICOS DE CARBAMATO Y CARBAZATO CETOLIDA.
(16/11/2005). Ver ilustración. Solicitante/s: PFIZER PRODUCTS INC.. Inventor/es: KANEKO, TAKUSHI-PFIZER INC., SU, WEI-GUO-PFIZER INC., WU, YONG-JIN-PFIZER INC.
Un compuesto de la fórmula o una sal farmacéuticamente aceptable, profármaco o solvato del mismo, en la que: X1 es CH2-o NR4; X2 es =O o =NOR1; Z es H, alquilo C1-C14, (aril C6-C10) (alquilo C1-C10)- o (heterociclo de 4-10 miembros) (alquilo C1-C10)-, donde uno o dos átomos de carbono de los radicales alquilo anteriores se reemplazan opcionalmente con un heteroátomo seleccionado entre O, S y -N(R4)-, y los grupos anteriores, excepto H, están opcionalmente sustituidos con 1 a 3 sustituyentes seleccionados independientemente entre halo, hidroxi, alcoxi C1-C14, alquilo C1-C14, (aril C6-C10) (alcoxi C1-C10)- y (heterociclo de 4-10 miembros) (alquilo C1-C10)-; R1 es H, metilo o etilo.
METODO PARA OBTENER CLARITROMICINA EN CRISTALES DE FORMA II.
(16/11/2005). Solicitante/s: HANMI PHARM. CO., LTD.. Inventor/es: SUH, KWEE, HYUN, SEONG, MI, RA, KIM, NAM, DU, LEE, GWAN, SUN, YUN, SANG-MIN, KIM, GI-JEONG.
Un método de obtención de la claritromicina en cristales de la forma II (fórmula I), que consiste en los pasos siguientes: (a) tratar una claritromicina de calidad no farma céutica con ácido metanosulfónico en una mezcla de un disol vente orgánico miscible con agua y agua para obtener el mesilato de claritromicina trihidratado cristalino de la fórmula (II); y (b) neutralizar el mesilato de claritromicina trihi dratado cristalino, obtenido en la etapa (a), con amoníaco acuoso en una mezcla de un disolvente orgánico miscible con agua y agua; con dicha claritromicina de calidad no farmacéutica se indica una claritromicina de cualquier pureza o de cualquier estadio de cristalinidad, incluido el producto en bruto obtenido a partir de un proceso de obtención de la misma:.
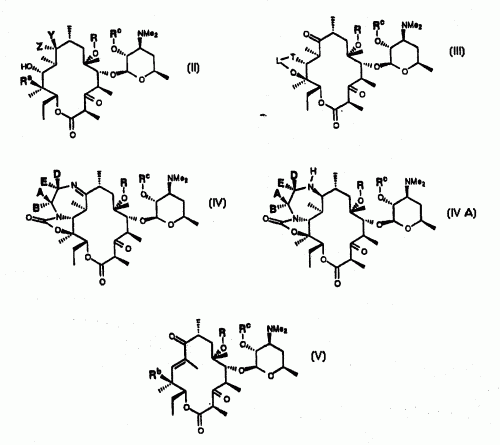
CETOLIDOS 6-0-SUSTITUIDOS QUE TIENEN ACTIVIDAD ANTIBACTERIANA.
(16/11/2005). Ver ilustración. Solicitante/s: ABBOTT LABORATORIES. Inventor/es: OR, YAT SUN, MA, ZHENKUN, CLARK, RICHARD, F., CHU, DANIEL, T., PLATTNER, JACOB, J..
Un compuesto con la siguiente fórmula (IV-A) o una sal farmacéuticamente aceptable del mismo, donde R p es hidrógeno o un grupo protector de hidroxi; R se selecciona del grupo formado por metilo sustituido con un radical seleccionado del grupo formado por (a) CN, (b) F, (c) S(O)nR 10 donde n es 0, 1 o 2 y donde R 10 es alquilo C1-C3 o alquilo C1-C3 sustituido con arilo o alquilo C1-C3 sustituido con heteroarilo, (d) NHC(O)R 10 donde R 10 se define como antes, (e) NHC(O)NR 11 R 12 donde R 11 y R 12 se seleccionan independientemente entre hidrógeno, alquilo C1-C3, alquilo C1-C3 sustituido con arilo, arilo sustituido, heteroarilo, heteroarilo sustituido, (f) arilo, (g) arilo sustituido, (h) heteroarilo, e (i) heteroarilo sustituido, alquilo C2-C10 sustituido con uno o más sustituyentes seleccionado del grupo formado por (a) halógeno, (b) hidroxi, (c) alcoxi C1-C3, (d) alcoxi C1-C3-alcoxi C1-C3, 5 10 (e) oxo, (f) -N3, (g) -CHO, (h) O-SO2-(alquilo C1-C6 sustituido), (i) -NR 13 R 14.
CETOLIDOS 6-0-SUSTITUIDOSQUE TIENEN ACTIVIDAD ANTIBACTERIANA.
(16/11/2005) Un compuesto con la siguiente fórmula (v) o una sal farmacéuticamente aceptable del mismo, donde Rb se selecciona del grupo formado por hidroxi, -O-C(O)-NH2 y -O-C(O)-imidazolilo; Rp es hidrógeno o un grupo protector de hidroxi; R se selecciona del grupo formado por metilo sustituido con un radical seleccionado del grupo formado por (a) CN, (b) F, (c) -CO2R10, donde R10 es alquilo C1-C3 o alquilo C1-C3 sustituido con arilo o alquilo C1-C3 sustituido con heteroarilo, (d) S(O)nR10 donde n es 0, 1 o 2 y R10 se define como antes, (e) NHC(O)R10 donde R10 se define como antes, (g) arilo, (h) arilo sustituido, (i) heteroarilo, y (j) heteroarilo sustituido, alquilo C2-C10 sustituido con uno o más sustituyentes seleccionado del grupo formado por (a) halógeno, (b) hidroxi, (c) alcoxi C1-C3, (d) alcoxi…
CETOLIDOS 6-0-SUSTITUIDOS QUE TIENEN UNA ACTIVIDAD ANTIBACTERIANA.
(16/11/2005). Ver ilustración. Solicitante/s: ABBOTT LABORATORIES. Inventor/es: OR, YAT SUN, MA, ZHENKUN, CLARK, RICHARD, F., CHU, DANIEL, T., PLATTNER, JACOB, J..
LA INVENCION SE REFIERE A COMPUESTOS ANTIMICROBIANOS DE FORMULAS (II), (III), ((V), (IV-A) O (V), ASI COMO A SUS SALES FARMACEUTICAMENTE ACEPTABLES, ESTERES O PROFARMACOS DE LOS MISMOS. SE DESCRIBEN ASIMISMO PROCEDIMIENTOS DE TRATAMIENTO DE INFECCIONES BACTERIANAS MEDIANTE LA ADMINISTRACION DE DICHOS COMPUESTOS, Y PROCEDIMIENTOS DE PREPARACION DE LOS MISMOS.
UTILIZACION DE UN AGENTE SUPRESOR DE LA INFECCION Y DE LA PROLIFERACION DEL VIH.
(01/11/2005). Ver ilustración. Solicitante/s: THE KITASATO INSTITITE. Inventor/es: OMURA, SATOSHI, SUNAZUKA, TOSHIAKI, AKAGAWA, KIYOKO.
Utilización de un derivado de macrólido en la fabricación de un medicamento para el tratamiento de un sujeto mediante la supresión de la infección y/o de la proliferación del virus del síndrome de la inmunodeficiencia humana en un macrófago derivado de un monocito humano, donde el derivado de macrólido es seleccionado de entre oxaciclotetradecano-2-10-diona , 4[2, 6- didesoxi-3-O-metil-á-L-ribo-hexopiranosil)oxi]-14-etil- 7, 12, 13-trihidroxi-3, 5, 7, 9, 11, 13-hexametil-6-[[3, 4, 6- tridesoxi-3-(dimetilamino)-â-D-xilo-hexopiranosil]oxi]; 11- (1'-hidroxipropil)-3-[2 , 6-didesoxi-3-C-metil-á-L-ribo- hexopiranosil]oxi]-5-[(3 , 4, 6-tridesoxi-3-(dimetilamino)-â-D- xilo-hexopiranosil)oxi]-2 , 4, 6, 8, 11, 14-hexametil-10, 13, 15-.
PROCEDIMIENTO PARA LA PREPARACION DE DERIVADOS DE ERITROMICINA 6-O-AL-QUENIL SUSTITUIDOS.
(01/11/2005). Ver ilustración. Solicitante/s: ABBOTT LABORATORIES. Inventor/es: WITTENBERGER, STEVEN, J., KING, STEVEN, A., HSU, MARGARET, CHI-PING, HAIGHT, ANTHONY, R., STONER, ERIC J., PETERSON, MATTHEW J., KU, YI-YIN, CINK, RUSSELL D., COOPER, ARTHUR J., DESHPANDE, MAHENDRA N., GRIEME, TIM, HILL, DAVID R., LEANNA, MARVIN R., LEE, ELAINE C., MCLAUGHLIN, MAUREEN A., MORTON, HOWARD E., NAPIER, JAMES J., PLATA, DANIEL J., RAJE, PRASAD S., RASMUSSEN, MICHAEL, RILEY, DAVID, TIEN, JIEN-HEH J..
Procedimiento para preparar derivados de eritromicina 6-O-sustituidos y cetólidos de eritromicina 6-O-sustituidos de los mismos. Específicamente, se refiere a un procedimiento catalizado por paladio para preparar derivados de eritromicina 6-O-sustituidos a partir de eritromicinas utilizando agentes alquilantes en presencia de una fosfina y su posterior conversión en cetólidos de eritromicina 6-O-sustituidos.
SALES DE ADICION DE AZITROMICINA Y ACIDO CITRICO Y PROCEDIMIENTO PARA SU OBTENCION.
(16/10/2005). Ver ilustración. Solicitante/s: QUIMICA SINTETICA, S.A.. Inventor/es: COSME GOMEZ,ANTONIO, PALOMO NICOLAU,FRANCISCO EUGE.
Sales de adición de azitromicina y ácido cítrico y procedimiento para su obtención. Dichas sales de adición tienen una relación molar entre azitromicina y ácido cítrico tal que proporciona un pH, en una disolución acuosa al 10%, comprendido entre 4,0 y 8,0. El procedimiento para preparar dichas sales comprende: a) disolver azitromicina en un disolvente o mezcla de disolventes, b) añadir ácido cítrico, y c) aislar el producto obtenido la sal por cristalización. Las sales de adición de azitromicina y ácido cítrico son estables y solubles en medio acuoso, siendo útiles agentes antibacterianos y antiprotozoarios.
ANTAGONISTAS MACROLIDAS DE LA HORMONA DE LIBERACION DE LA LUTEINOESTIMULINA (LHRH).
(01/09/2005) Un compuesto representado por la fórmula: o una sal farmacéuticamente aceptable o éster del mismo, donde A se selecciona entre el grupo compuesto por: (a) -C, (b) -N, y (c) -O; X e Y se seleccionan independientemente en cada aparición entre el grupo compuesto por: (a) hidrógeno, (b) haluro, (c) trifluorometilo, (d) alcoxi, (e) alquilo, (f) arilo, y (g) arilo sustituido; R se selecciona entre el grupo compuesto por: (a) alquilo, (b) cicloalquilo, (c) cicloalquilo sustituido, (d) heterociclilo, (e) heterociclilo sustituido, (f) cicloalquilalquilo, (g) cicloalquilalquilo sustituido, (h) alquilarilo, (i) alquilheterociclilo, (j) alquenilo, (k) alquinilo, (l) -C(S)-NHR4, C(NR4)-NHR4,…
PROCEDIMIENTO PARA PREPARACION DE POLIMORFOS DE CLARITROMICINA.
(16/07/2005). Solicitante/s: TEVA PHARMACEUTICAL INDUSTRIES LTD.. Inventor/es: LIFSHITZ, IGOR, AVRUTOV, ILYA, SCHWARTZ, EDI, MASARWA, BASEM.
Procedimiento para convertir la Forma I polimórfica de la claritromicina en la Forma II polimórfica de la claritromicina, que comprende suspender un sólido que comprende la Forma I de la claritromicina en agua.
SAL DIFOSFATO DE UN DERIVADO DE 4'-SUSTITUIDO-9- DESOSXO-9A-HOMOERITROMICINA Y SU COMPOSICION FARMACEUTICA.
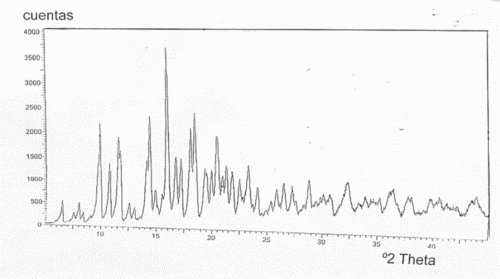
(16/06/2005). Ver ilustración. Solicitante/s: PFIZER PRODUCTS INC.. Inventor/es: RAFKA, ROBERT JOHN, PFIZER INC., CENTRAL RES. DIV., RAGAN, COLMAN BRENDAN, PFIZER INC., CTRL RES. DIV., ALLEN, DOUGLAS JOHN MELDRUM, PFIZER INC.
Dos polimorfos del difosfato de (2R, 3S, 4R, 5R, 8R, 10R, 11R, 12S, 13S, 14R)-13-[[2, 6-didesoxi-3- C-metil-3-0-metil-4-C-[(propilamino)metil]-á-L-ribo- hexopiranosil]oxi]-2-etil-3 , 4, 10-trihidroxi- 3, 5, 8, 10, 12, 14-hexametil-11-[[3, 4, 6-tridesoxi-3- (dimetilamino)-ß-D-xilo-hexopiranosil]oxi]-1-oxa-6- azaciclopentadecan-15-ona de fórmula: 4 en la que n es de 0 a 8 y los polimorfos se seleccionan entre el grupo constituido por: a. una mesofase cristalina esméctica de un cristal líquido; y b. un difosfato cristalino que presenta el modelo de difracción de rayos X de polvo Nº pico 1 2 3 4 5 6 7 8 9 10 Espacio 16, 12, 4 10, 8 9, 0 6, 9 6, 5 6, 2 5, 4 5, 1 4, 9 d 2.
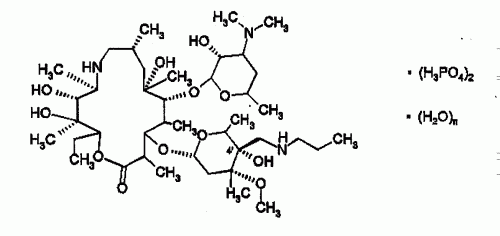
METODO PARA PREPARAR UN AZA-MACROLIDO CON 4" (R)-NH2.
(01/06/2005). Ver ilustración. Solicitante/s: MERIAL. Inventor/es: LEON, PATRICK, LHERMITTE, FREDERIC, ODDON, GILLES, GUEVEL, RONAN, PAUZE, DENIS, GAREL, LAURENT.
Procedimiento de preparación estereoselectivo de un compuesto de fórmula general I **(Fórmula)** donde: - R es un átomo de hidrógeno o un grupo alquilo C1-C10, alquenilo C2-C10 o arilsulfonilo C6-C12, según sea el caso substituidos, y - los A, idénticos o diferentes, representan un átomo de hidrógeno, un átomo de nitrógeno, según sea el caso substituido, un grupo alquilo C1-C4 eventualmente substituido por uno o varios grupos arilo, según sea el caso substituidos, un grupo R2CO o R2SO2, representando R2 un átomo de hidrógeno, un grupo alquilo C1-C8 o arilo, según sea el caso substituidos, y - el símbolo V indica que ha habido una inversión de configuración a nivel del carbono C-4 con respecto al compuesto de fórmula general II, a partir de un compuesto de fórmula general II.
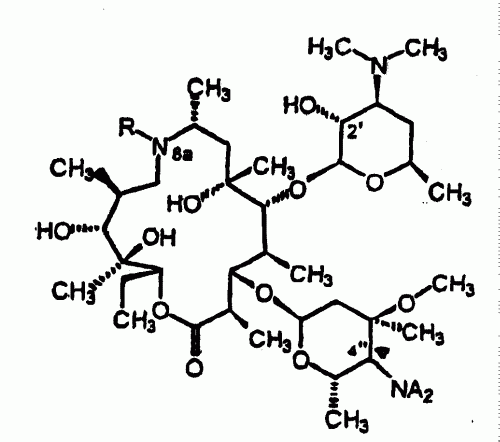
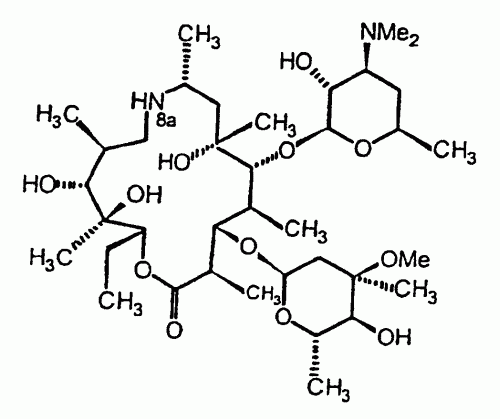
PROCEDIMIENTO PARA LA PREPARACION DE DERIVADOS DE 9-DEOXO-8A-AZA-(8A-ALQUIL)-8A-HOMOERITROMICINA A A PARTIR DE LA 9-DEOXO-9 (Z)-HIDROXIIMINOERITROMICINA A.
(01/06/2005). Ver ilustración. Solicitante/s: MERIAL. Inventor/es: LEON, PATRICK, LHERMITTE, FREDERIC, ODDON, GILLES, PAUZE, DENIS.
Procedimiento de preparación de la 9-deoxo-8a-aza-8a- homoeritromicina A de fórmula V a través del reordenamiento estereoespecífico de Beckmann en un medio de reacción que utiliza la piridina como disolvente principal, de un compuesto de fórmula II en dos intermediarios imidatos III y IV y la posterior reducción de dichos compuestos III y IV, caracterizado porque dichos compuestos III y IV formados en el medio de reacción del reordenamiento de Beckmann no están aislados de dicho medio y están directamente implicados en la etapa de reducción mediante una cantidad suficiente de borohidruro, previa extracción de la piridina mediante un hidrocarburo miscible con ésta y en el que dichos imidatos III y IV, en forma de sal, son insolubles.
PROCESO PARA LA PREPARACION DE 6-0-METIL-ERITROMICINA A USANDO DERIVADOS DE 9-HIDROXI ERITROMICINA.
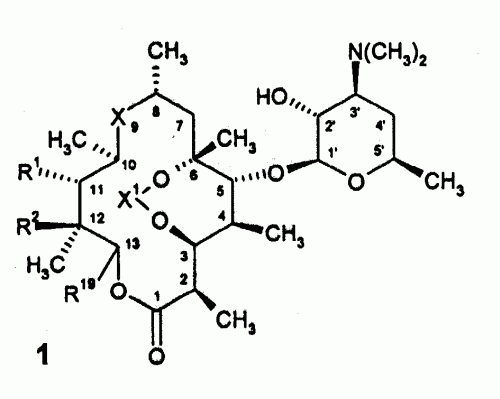
(16/04/2005). Solicitante/s: ABBOTT LABORATORIES. Inventor/es: KU, YI-YIN, LEE, ELAINE C., TIEN, JIEN-HEH J., RILEY, DAVID A..
Un proceso para la preparación de 6-O-metil- eritromicina A que comprende: a) Proteger el grupo 2-hidroxilo de la eritromicina A para formar un derivado de eritromicina A protegido en 2; b) Reducir el grupo 9-ceto del derivado de eritromicina A protegido en 2 para formar un derivado de 9-hidroxi- eritromicina A protegido en 2; c) Proteger la 9-hidroxi eritromicina A protegida en 2 para formar un derivado de eritromicina A protegido en las posiciones 9 y en 2; d) Metilar la posición 6 del derivado de eritromicina A protegido en las posiciones 9 y en 2 para formar un derivado de 6-O-metil-eritromicina A protegido en las posiciones 9 y en 2. e) Desproteger el derivado de 6-O-metil-eritromicina A protegido en las posiciones 9 y 2 para formar un derivado de 9-hidroxi de eritromicina A 6-O-metilado; y f) Oxidar el 9-hidroxi de la 9-hidroxi eritromicina A 6-O- metilada para formar 6-O-metil-eritromicina A.
DERIVADOS DE 6-0-ALQUILO ERITROMICINA B.
(16/04/2005). Ver ilustración. Solicitante/s: ABBOTT LABORATORIES. Inventor/es: MONTGOMERY, STEPHEN, H., LIU, JIH-HUA.
Un compuesto de estructura I, tal como se representa a continuación: en el que R1 es alquilo, R2 y R4 son cada uno de ellos independientemente hidrógeno o un grupo O-protector convencional como sililo, alquilcarbonilo, alcoxicarbonilo, acilo, alquenil monocarbonilo inferior, alcoxicarbonilalquilcarbonilo inferior o arilcarbonilo; y R3 es NR5(CH3), donde R5 es metilo (CH3) o un grupo N-protector convencional, o R3 es N+(CH3)2R6X-, donde R6 es 2-aquenilo, bencilo o bencilo sustituido y X es un halógeno.
(16/04/2005). Ver ilustración. Solicitante/s: DOW AGROSCIENCES LLC ELI LILLY AND COMPANY. Inventor/es: LEWER, PAUL, HAHN, DONALD, R., KARR, LAURA, L., GRAUPNER, PAUL, R., GILBERT, JEFFREY, R., WORDEN, THOMAS, V., YAO, RAYMOND, C., NORTON, DENNIS, W.
Un compuesto de la fórmula ó : **(Fórmulas)** o una de sus sales, en la que R1 es un grupo de fórmula 2a, 2b, o 2c **(Fórmulas)** R2 es H u OH; R3 es H o CH3 R4 en la fórmula 1 es 1-butenil, 1, 3-butadienil, n-butil, 3-hidroxi-1-butenil, o 1-propenil; y R4 es etilo en la fórmula 55 y R5 es H o un grupo que tiene una de las siguientes fórmulas 4a a 4i **(Fórmulas)**.
ETANNOLATO DE AZITROMICINA, PROCEDIMIENTO PARA SU FABRICACION Y COMPOSICIONES FARMACEUTICAS QUE LO CONTIENEN.
(16/04/2005). Ver ilustración. Solicitante/s: TEVA PHARMACEUTICAL INDUSTRIES LTD.. Inventor/es: ARONHIME, JUDITH, SINGER, CLAUDE.
Etanolato de azitromicina con un contenido en etanol comprendido entre aproximadamente el 1, 5% y aproximadamente el 3%.
ANTIBIOTICOS MACRILIDOS DE 3,6-CETAL.
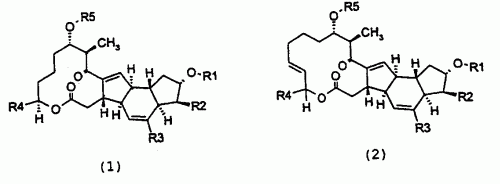
(16/03/2005). Ver ilustración. Solicitante/s: PFIZER PRODUCTS INC.. Inventor/es: CHENG, HENGMIAO, LUNDY, KRISTIN M., SAKYA, SUBAS M., BERTINATO, PETER, MINICH, MARTHA L.
LA INVENCION SE REFIERE A COMPUESTOS DE FORMULAS 1 Y 2: Y A SALES Y SOLVATOS FARMACEUTICAMENTE ACEPTABLES DE LOS MISMOS, DONDE X, X 1 , R 1 , R 2 , R 7 , R 17 Y R19 SON TAL Y COMO SE LOS DEFINE EN LA DESCRIPCION. LOS C OMPUESTOS DE FORMULAS 1 Y 2 PUEDEN SER UTILES EN EL TRATAMIENTO DE INFECCIONES BACTERIANAS, PARASITARIAS Y PROTOZOARIAS, ASI COMO EN TRANSTORNOS RELACIONADOS CON DICHAS INFECCIONES, EN MAMIFEROS, PECES Y AVES. LA INVENCION SE REFIERE ASIMISMO A COMPOSICIONES FARMACEUTICAS QUE CONTIENEN DICHOS COMPUESTOS DE FORMULAS 1 Y 2 Y A METODOS DE TRATAMIENTO DE INFECCIONES BACTERIANAS, PARASITARIAS Y PROTOZOARIAS, MEDIANTE LA ADMINISTRACION DE DICHOS COMPUESTOS.
NUEVOS DERIVADOS DE MACROLIDOS.
(16/03/2005). Solicitante/s: PFIZER PRODUCTS INC.. Inventor/es: LETAVIC, MICHAEL, ANTHONY, BRONK, BRIAN SCOTT, PFIZER INC., CENTRAL RES. DIV., CHENG, HENGMIAO, PFIZER INC., CENTRAL RES. DIV., DUTRA, JASON KENNETH, PFIZER INC., CTRL. RES. DIV., RAFKA, ROBERT JOHN, PFIZER INC., CTRL. RES. DIV.
La invención se refiere a nuevos compuestos de fórmula Iy a las sales farmacéuticamente aceptables de éstos, en la que R{sup,1}, R{sup,2}, R{sup,3}, Q, X, Y y Z son como se definen en la descripción. La invención también se refiere a compuestos farmacéuticos que contienen a los compuestos de fórmula I, a los procedimientos de empleo de dichos compuestos de fórmula I para el tratamiento de infecciones y a los procedimientos para preparar a dichos compuestos de fórmula I.
UN PROCEDIMIENTO PARA LA OBTENCION DE CLARITROMICINA.
(01/03/2005). Solicitante/s: ERCROS INDUSTRIAL, S.A.. Inventor/es: DIAZ TEJO,LUIS ANGEL, BORRELL BILBAO,JOSE IGNACIO, ASENSIO COMINGUEZ,RAMON, CRUZADO RODRIGUEZ,M. DEL CARMEN, NOMEN RIBE,ROSA, SEMPERE CEBRIAN,JULIA.
Un procedimiento para la obtención de claritromicina. Este procedimiento está destinado a la obtención de la claritromicina; según el procedimiento se parte del clorohidrato de 9-oxima de eritromicina A, el cual se transforma en claritromicina mediante una secuencia sintética en la que inicialmente se forma un acetal de la 9-oxima. El uso del clorohidrato de oxima permite que sólo sea necesario utilizar cantidades catalíticas de sales de piridina para favorecer la reacción. Seguidamente se protegen los hidroxilos deposiciones 2' y 4'' con un agente sililante, se metila el hidroxilo deposición 6; todo ello sin que sea necesario el aislamiento de ningún intermedio de reacción. Finalmente la desprotección del acetal y de los silanos de 2' y 4'', seguida de la desoximación rinde claritromicina con alto rendimiento y de forma fácilmente aplicable industrialmente.
SAPONINAS ANTIPROTOZOARIAS.
(01/03/2005) Una saponina triterpénica que se puede obtener mediante un proceso para el aislamiento de saponinas triterpénicas que se encuentran en la familia Myrsinaceae donde las saponinas se aislan de la planta de la especie Maesa balansae y donde dicho proceso comprende los pasos de (a) extracción de las partes de la planta seca con un alcohol y concentración del extracto, (b) eliminación de la fracción apolar del extracto mediante extracción líquido-líquido con un disolvente apolar, (c) purificación posterior de las saponinas del extracto alcohólico mediante extracción líquido-líquido, filtración y cromatografía, y (d) la cromatografía comprende…
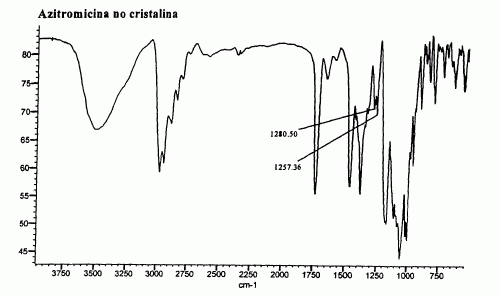
PREPARACION DE AZITROMICINA EN SU FORMA DIHIDRATO CRISTALINA.
(01/03/2005). Ver ilustración. Solicitante/s: ASTUR PHARMA, S.A. Inventor/es: LLORENTE GARCIA,ISIDRO, BAYOD JASANADA,MIGUEL SANTOS, FERNANDEZ MARI,FELIX.
La invención describe un procedimiento en el que el producto final se obtiene por cristalización desde una solución de azitromicina en Terc.Butanol, bien por adición de agua, bien por adición de esa disolución sobre una mezcla de eter de petroleo/agua. La invención describe métodos analíticos para diferenciar las formas no cristalina y la dihidrato cristalizada de azitromicina.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}