Derivados antibióticos de binaftilo.
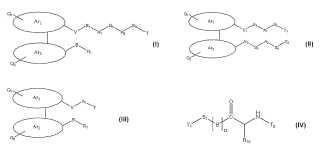
Un compuesto de fórmula Ia, **Fórmula**
o una sal farmacéuticamente aceptable o hidrato del mismo,
donde:
cada uno de Q1 y Q2 se selecciona independientemente entre hidrógeno, alquilo C1-C12, cicloalquilo C3-C6,alquiloxi C1-C12, nitro, halógeno, hidroxilo, amino, mono o dialquilamino, ácido carboxílico o una sal o éster delmismo, ácido sulfónico o una sal o éster del mismo, ácido fosfórico o una sal o éster del mismo, o un grupo quecontiene nitrógeno, tal como carboxamida, sulfonamida o fosforamida, donde cada alquilo C1-C12, alquiloxi C1-C12o cicloalquilo C3-C6 está opcionalmente sustituido con hidroxilo, amino, ácido carboxílico o una sal o éster delmismo, ácido sulfónico o una sal o éster del mismo, ácido fosfórico o una sal o éster del mismo, o un grupo quecontiene nitrógeno tal como carboxamida, sulfonamida o fosforamida;
B se selecciona entre -O-, -S-, -S(O)-, -S(O)2-, -NH-, y -N(alquilo C1-C6)-;
R1 se selecciona entre hidrógeno, alquilo C1-C12, alquil C1-C6-cicloalquilo C3-C6, alquil C1-C6-arilo C6-C10,alquenilo C2-C6, alquinilo C2-C6, un polioxialquileno que tiene de 2 a 6 átomos de carbono, y cuando B es -S-, -S(O)-, -S(O)2-, -NH- o -N(alquil C1-C6)- entonces R1 puede ser hidroxilo;
V es un grupo enlazador seleccionado entre -O-, -O-L-C(O), -O-L-NR6-, -C(O)-, -NR6-, -S(O)-, -S(O)2-, -O-L-S(O)-, -S(O)2-L- C(O)-, -S(O)2-L-NR6-, P(O)2O-;
donde L se selecciona entre alquilo C1-C12, alquenilo C2-C8, cicloalquilo C3-C6, polioxialquileno que tiene de 2 a 6átomos de carbono, arilo C6-C10 y alquil C1-C6-arilo C6-C10 y donde R6 se selecciona entre H, alquilo C1-C12;cada uno de A1 y A2 se selecciona independientemente entre el grupo que consiste en lisina, arginina y ornitina;cada uno de S1, S2 y S3 está presente o ausente y es un resto α- o β-aminoacídico seleccionadoindependientemente;
T está presente o ausente y se selecciona entre -C(O)OR8, -OR8, -NHR8, NHOR8, -NH-aril C6-CO-R8, -NH-aril C6-CO-NHR8, -NH-aril C6-CONHOR8, -NH-aril C6-CONHOH, - C(O)NHR8, -(NH)-SO2arilo C6, -(NH)COR8;o T forma un isóstero de carboxilato, opcionalmente sustituido con R8, que reemplaza al grupo ácido carboxílicodel aminoácido al que está conectado T;
donde dicho isóstero de carboxilato es un tetrazol, isoxazol, oxazol o tiazol;
donde R8 se selecciona entre hidrógeno, alquilo C1-C12, alquil C1-C6-arilo C6-C10, alquil C1-C6-cicloalquilo C3-C6,alquenilo C2-C6 y alquinilo C2-C6; y
donde, cuando T está conectado con el extremo C de un resto aminoacídico, entonces el grupo carbonilo delresto aminoacídico puede estar reducido para dar metileno.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/AU2005/001444.
Solicitante: UNIVERSITY OF WOLLONGONG.
Nacionalidad solicitante: Australia.
Dirección: NORTHFIELDS AVENUE WOLLONGONG, NSW 2522 AUSTRALIA.
Inventor/es: BRKIC, ZINKA, COATES,Jonathan,Alan,Victor, RHODES,David,Ian, BREMNER,John,Barnard, BOYLE,TIMOTHY PATRICK, DALTON,NEAL KEVIN, DEADMAN,JOHN, KELLER,PAUL ANTHONY, MORGAN,JODY, PYNE,STEPHEN GEOFFREY, ROBERTSON,MARK JAMES.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A61K31/165 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › teniendo ciclos aromáticos, p. ej. colchicina, atenolol, progabide.
- A61K31/167 A61K 31/00 […] › teniendo el átomo de nitrogeno de un grupo carboxiamida unido directamente al ciclo aromático, p. ej. lidocaina, paracetamol.
- A61K31/216 A61K 31/00 […] › de ácidos que tienen ciclos aromáticos, p. ej. benacticina, clofibrato.
- A61K31/245 A61K 31/00 […] › del tipo ácido aminobenzoico, p. ej. procaína, novocaína (ésteres del ácido salicílico A61K 31/60).
- A61K31/353 A61K 31/00 […] › 3,4-Dihidrobenzopiranos, p. ej. cromano, catequina.
- A61K31/395 A61K 31/00 […] › que tienen el nitrógeno como heteroátomo de un ciclo, p. ej. guanetidina o rifamicina.
- A61K31/421 A61K 31/00 […] › 1,3-Oxazoles, p. ej. pemolina, trimetadiona.
- A61K31/4402 A61K 31/00 […] › sustituidos unicamente en posición 2, p. ej. feniramina, bisacodil.
- A61P31/04 A61 […] › A61P ACTIVIDAD TERAPEUTICA ESPECIFICA DE COMPUESTOS QUIMICOS O DE PREPARACIONES MEDICINALES. › A61P 31/00 Antiinfecciosos, es decir antibióticos, antisépticos, quimioterápicos. › Agentes antibacterianos.
- A61P31/18 A61P 31/00 […] › para el VIH.
- C07C231/02 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07C COMPUESTOS ACICLICOS O CARBOCICLICOS (compuestos macromoleculares C08; producción de compuestos orgánicos por electrolisiso electroforesis C25B 3/00, C25B 7/00). › C07C 231/00 Preparación de amidas de ácidos carboxílicos. › a partir de ácidos carboxílicos o a partir de sus ésteres, anhídridos o haluros por reacción con amoniaco o aminas.
- C07C233/47 C07C […] › C07C 233/00 Amidas de ácidos carboxílicos. › con el átomo de carbono del grupo carboxamido unido a un átomo de hidrógeno o a un átomo de carbono de una estructura carbonada acíclica saturada.
- C07C233/48 C07C 233/00 […] › con el átomo de carbono del grupo carboxamido unido a un átomo de carbono acíclico de una estructura carbonada saturada que contiene ciclos.
- C07C233/49 C07C 233/00 […] › con el átomo de carbono del grupo carboxamido unido a un átomo de carbono de una estructura carbonada acíclica insaturada.
- C07C233/54 C07C 233/00 […] › con el átomo de carbono del grupo carboxamido unido a un átomo de hidrógeno o a un átomo de carbono de una estructura carbonada saturada.
- C07C233/64 C07C 233/00 […] › con átomos de carbono de grupos carboxamido unidos a átomos de carbono de ciclos aromáticos de seis miembros.
- C07C233/78 C07C 233/00 […] › con el radical hidrocarbonado sustituido unido al átomo de nitrógeno del grupo carboxamido por un átomo de carbono acíclico.
- C07C235/26 C07C […] › C07C 235/00 Amidas de ácidos carboxílicos, estando sustituida la estructura carbonada de la parte ácida por átomos de oxígeno. › siendo saturada la estructura carbonada y conteniendo ciclos.
- C07C235/30 C07C 235/00 […] › siendo la estructura carbonada insaturada y conteniendo ciclos distintos de los ciclos aromáticos de seis miembros.
- C07C235/34 C07C 235/00 […] › con los átomos de nitrógeno de los grupos carboxamido unidos a átomos de hidrógeno o a átomos de carbono acíclicos.
- C07C269/06 C07C […] › C07C 269/00 Preparación de derivados del ácido carbámico, es decir, de compuestos que contienen uno de los grupos en que el átomo de nitrógeno no forma parte de grupos nitro o nitroso. › por reacciones que no implican la formación de grupos carbamato.
- C07C271/00 C07C […] › Derivados del ácido carbámico, es decir, compuestos que contienen uno de los grupos en que el átomo de nitrógeno no forma parte de grupos nitro o nitroso.
- C07C277/00 C07C […] › Preparación de guanidina o sus derivados, es decir, de compuestos que contienen el grupo en que los átomos de nitrógeno unidos por enlaces sencillos no forman parte de grupos nitro o nitroso.
- C07C279/04 C07C […] › C07C 279/00 Derivados de guanidina, es decir, compuestos que contienen el grupo en que los átomos de nitrógeno unidos por enlaces sencillos no forman parte de grupos nitro o nitroso. › que tienen átomos de nitrógeno de grupos guanidina unidos a átomos de carbono acíclicos de una estructura carbonada.
- C07C279/24 C07C 279/00 […] › siendo Y un heteroátomo.
- C07D213/46 C07 […] › C07D COMPUESTOS HETEROCICLICOS (Compuestos macromoleculares C08). › C07D 213/00 Compuestos heterocíclicos que contienen ciclos de seis miembros, no condensados con otros ciclos, con un átomo de nitrógeno como el único heteroátomo del ciclo y tres o más enlaces dobles entre miembros cíclicos o entre miembros cíclicos y miembros no cíclicos. › Atomos de oxígeno.
- C07D263/32 C07D […] › C07D 263/00 Compuestos heterocíclicos que contienen ciclos de oxazol-1,3 u oxazol-1,3 hidrogenado. › con solamente átomos de hidrógeno, radicales hidrocarbonados o hidrocarbonados sustituidos, unidos directamente a los átomos de carbono del ciclo.
- C07D263/46 C07D 263/00 […] › Atomos de azufre.
- C07D273/02 C07D […] › C07D 273/00 Compuestos heterocíclicos que contienen ciclos que tienen átomos de nitrógeno y oxígeno como únicos heteroátomos del ciclo, no previstos por los grupos comprendidos entre el C07D 261/00 - C07D 271/00. › que tienen dos átomos de nitrógeno y un solo átomo de oxígeno.
- C07D273/08 C07D 273/00 […] › que tienen dos átomos de nitrógeno y varios átomos de oxígeno.
- C07D311/70 C07D […] › C07D 311/00 Compuestos heterocíclicos que contienen ciclos de seis miembros que contienen un átomo de oxígeno como único heteroátomo, condensados con otros ciclos. › con dos radicales hidrocarbonados unidos en la posición 2 y elementos diferentes al hidrógeno y al carbono en la posición 6.
PDF original: ES-2412483_T3.pdf
Fragmento de la descripción:
Derivados antibióticos de binaftilo Campo de la invención Esta invención se refiere a nuevos compuestos peptídicos, a métodos para prepararlos y a su uso como antibióticos y en el tratamiento de infecciones por VIH.
Antecedentes de la invención Las bacterias y las infecciones bacterianas que pueden tratarse por antibióticos incluyen, aunque sin limitación las siguientes:
Staphylococcus aureus, (o "staph") , son bacterias halladas habitualmente en la piel y en la nariz de gente sana, (F) y son una de las causas principales de infecciones cutáneas y también pueden causar infecciones graves y a veces fatales (tales como infecciones del torrente sanguíneo incluyendo síndrome por shock tóxico, impétigo, infecciones en heridas quirúrgicas, infecciones de implantes plásticos, osteomielitis y neumonía) .
Enterococos, que se han conocido como una causa de endocarditis infecciosa durante casi un siglo, más recientemente también se han reconocido como causa de infección nosocomial y "superinfección" en pacientes que reciben agentes antimicrobianos.
Otras bacterias Gram positivas que pueden tratarse por antibióticos incluyen Staphylococcus epidermitis que causa endocarditis, Clostridium difficile que causa diarrea y colitis pseudomembranosa, Bacillus anthracis de ántrax y Streptococcus pneumoniae que causa neumonía, meningitis, septicemia y otitis media en niños (o dolor de oído) . La familia de bacterias Streptococcus también puede dividirse en grupo A o Pyrogenes, que está implicado en envenenamiento de la sangre, glomerulonefritis y fiebres, tales como fiebre puerperal, escarlatina y reumática. El grupo B o Streptococcus agalactiae causa meningitis en el neonato y neumonía.
Resistencia bacteriana a antibióticos Las infecciones bacterianas pueden suceder mientras se está en el hospital (nosocomiales) , pero un problema adicional es el aumento de infecciones que se adquieren mientras la persona está en la comunidad. Un estudio reciente (C) identificó el perfil de susceptibilidad antimicrobiana y los mecanismos de resistencia de aislados MRSA pre-tratamiento obtenidos de sujetos adultos que participaban en ensayos recientes de tratamiento clínico de infecciones respiratorias adquiridas en la comunidad. De los 465 aislados de S. aureus, 43 se identificaron como MRSA. El ensayo de la susceptibilidad antimicrobiana indicó tasas de susceptibilidad a: vancomicina (100%) , gentamicina (86%) , clindamicina (39%) , quinolonas (49%) , y eritromicina (12%) . Todos los aislados resistentes a ciprofloxacina tenían un cambio de aminoácido en GyrA y GrlA. Los resultados indican que MRSA de sujetos adultos con infecciones respiratorias adquiridas en la comunidad tienen perfiles de susceptibilidad antimicrobiana y mecanismos de resistencia similares a MRSA nosocomiales.
El potencial patogénico de Staphylococcus aureus en infecciones nosocomiales y adquiridas en la comunidad es bien conocido. Cuando se introdujo la penicilina a mediados de la década de 1940, S. aureus era casi un 94% susceptible a este fármaco. Desarrolló resistencia generalizada a la penicilina en la década de 1950, seguido de resistencia a penicilinas semi-sintéticas en la década de 1960 y 1970. Desde entonces, se han propagado por todo el mundo cepas de S. aureus resistentes a meticilina y estafilococos coagulasa negativos resistentes a meticilina. La prevalencia de S. aureus resistente a meticilina varía geográficamente. En Argentina alcanza casi el 50%. La resistencia a meticilina en estafilococos se desarrolla debido a la proteína de unión a penicilina adicional PBP2a codificada por el gen mecA y es un problema grave tanto para los microbiólogos como para los médicos (A) . La alta prevalencia de estafilococos resistentes a meticilina compromete el uso de penicilinas semi-sintéticas para tratamientos clínicos en muchas instituciones, aumentando de este modo el uso de vancomicina (un glucopéptido) . Hasta 1996, los glucopéptidos eran casi universalmente activos contra S. aureus pero entonces se describió el primer S. aureus de glucopéptido intermedio (GISA) también conocido como VISA (S. aureus resistente de forma intermedia a vancomicina) y se aisló en Japón, seguido por Francia y Estados Unidos. Sigue informándose de infecciones con Staphylococcus aureus con susceptibilidad reducida a vancomicina, incluyendo dos casos causados por aislados de S. aureus con resistencia completa a vancomicina. (A) También existe S. aureus resistente a vancomicina (VRSA) . El aumento mundial en la incidencia de aislados clínicos de S. aureus con susceptibilidad reducida a vancomicina y teicoplanina indica que la resistencia a glucopéptidos en S. aureus se está convirtiendo en un problema clínico importante.
Los mecanismos exactos implicados no se han dilucidado aún, aunque VISA está asociado con síntesis aumentada de la pared. Muchas cepas VISA se caracterizan por biosíntesis aumentada de la pared celular y entrecruzamiento disminuido de las cadenas laterales peptídicas, conduciendo a la acumulación de extremos D-alanil-D-alanina libres en el peptidoglucano, que se ha propuesto que pueden actuar como sitios diana falsos para vancomicina. (B)
El mecanismo de resistencia a vancomicina en enterococos está bien definido y parece ser diferente al de VISA.
La resistencia a vancomicina en enterococos, conocidos como VRE o enterococos resistentes a glucopéptidos (GRE) , existe como resistencia intrínseca en que aislados de Enterococcus gallinarum y E. casseliflavus/E. flavescens muestran una resistencia inherente de bajo nivel a vancomicina o por resistencia adquirida en que los enterococos se vuelven resistentes a vancomicina por adquisición de información genética de otro organismo. Más frecuentemente, esta resistencia se observa en E. faecilum y E. faecalis, pero también se ha reconocido en E. raffinosus, E. avium, E. durans, y otras varias especies de enterococos.
Varios genes, incluyendo vanA, vanB, vanC, vanD, y vanE, contribuyen a la resistencia a vancomicina en enterococos. E. faecium es la especie más frecuentemente aislada de VRE en hospitales y típicamente produce concentraciones inhibidoras mínimas (MIC) elevadas a vancomicina (>128 !g/ml) y teicoplanina (>16 !g/ml) . Estos aislados típicamente contienen genes vanA. La epidemiología de Enterococcus faecium resistente a vancomicina (VREF) en Europa está caracterizada por una gran reserva en la comunidad. En contraste, los brotes e infecciones nosocomiales (sin una reserva en la comunidad) caracterizan a VREF en los Estados Unidos. (G)
En enterococos susceptibles a vancomicina, se añade D-alanil-D-alanina (formado por una D-alanina-D-alanina ligasa endógena) a un precursor tripéptido para formar un precursor pentapéptido. El extremo D-Ala-D-Ala es la diana de vancomicina; una vez se ha unido vancomicina, se evita el uso de este precursor pentapéptido para síntesis adicional de pared celular. En el fenotipo VanA, una de las proteínas cuya síntesis se induce por exposición a células bacterianas a vancomicina se llama VanA; VanA es una ligasa y se parece a la D-alanina-D-alanina ligasa de Escherichia coli y otros organismos, incluyendo enterococos susceptibles a vancomicina. VanA genera D-Ala-D-X, donde X es habitualmente lactato; la formación de lactato se debe a la presencia de VanH, una deshidrogenasa codifica por VanH. El resto depsipéptido, D-Ala-D-Lac, después se añade a un precursor tripéptido, produciendo un precursor depsipentapéptido. La vancomicina no se une al extremo D-Ala-D-Lac, de modo que este depsipentapéptido puede usarse en las etapas restantes de síntesis de pared celular. Sin embargo, cuando el precursor pentapéptido normal que finaliza en D-Ala-D-Ala también está presente, las células no son completamente resistentes a vancomicina, a pesar de la presencia de precursores que contienen D-Ala-D-Lac. Este aparente problema está controlado en gran parte por vanX, que codifica una dipeptidasa, VanX, que escinde D-Ala-D-Ala, evitando su adición al precursor tripéptido. Si algún D-Ala-D-Ala escapa de la escisión y provoca un precursor pentapéptido normal, vanY codifica una función complementaria o de apoyo. Es decir, codifica una carboxipeptidasa, VanY, que escinde la D-alanina y el D-lactato de extremos D-Ala-D-Ala y D-Ala-D-Lac, respectivamente, produciendo precursores tetrapéptidos, a los que no se une la vancomicina. Los otros genes implicados en el complejo de resistencia VanA incluyen VanR y VanS, cuyas proteínas codificadas están implicadas en la detección de la presencia de vancomicina extracelular o su efecto y señalización intracelular para activar la transcripción de vanH, vanA, y vanX. Un gen final en el grupo vanA es vanZ, que codifica VanZ, cuya tarea es desconocida. (J)
VanB, codificada... [Seguir leyendo]
Reivindicaciones:
1. Un compuesto de fórmula Ia,
o una sal farmacéuticamente aceptable o hidrato del mismo, donde:
cada uno de Q1 y Q2 se selecciona independientemente entre hidrógeno, alquilo C1-C12, cicloalquilo C3-C6, alquiloxi C1-C12, nitro, halógeno, hidroxilo, amino, mono o dialquilamino, ácido carboxílico o una sal o éster del mismo, ácido sulfónico o una sal o éster del mismo, ácido fosfórico o una sal o éster del mismo, o un grupo que contiene nitrógeno, tal como carboxamida, sulfonamida o fosforamida, donde cada alquilo C1-C12, alquiloxi C1-C12
2. Un compuesto de acuerdo con la reivindicación 1 donde A1 se selecciona entre lisina y ornitina y A2 se selecciona entre arginina.
3. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 2 donde S1 y S2 están ausentes.
4. Un compuesto de fórmula II,
o una sal farmacéuticamente aceptable o hidrato del mismo, donde:
Ar1-Ar2 es 1, 1'-binaftilo 2, 2'-sustituido; cada uno de Q1 y Q2 se selecciona independientemente entre hidrógeno, alquilo C1-C12, cicloalquilo C3-C6, alquiloxi C1-C12, nitro, halógeno, hidroxilo, amino, mono o dialquilamino, ácido carboxílico o una sal o éster del mismo, ácido sulfónico o una sal o éster del mismo, ácido fosfórico o una sal o éster del mismo, o un grupo que contiene nitrógeno tal como carboxamida, sulfonamida o fosforamida, donde cada alquilo C1-C12, alquiloxi C1-C12
5. Un compuesto de acuerdo con la reivindicación 4 donde A1 y A2 son iguales.
6. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 4 donde S1, S2, S3 y S4 están ausentes.
7. Un compuesto de fórmula III,
o una sal farmacéuticamente aceptable o hidrato del mismo, donde:
Ar1-Ar2 es 1, 1'-binaftilo 2, 2'-sustituido; cada uno de Q1 y Q2 se selecciona independientemente entre hidrógeno, alquilo C1-C12, cicloalquilo C3-C6, alquiloxi C1-C12, nitro, halógeno, hidroxilo, amino, mono o dialquilamino, ácido carboxílico o una sal o éster del mismo, ácido sulfónico o una sal o éster del mismo, ácido fosfórico o una sal o éster del mismo, o un grupo que contiene nitrógeno, tal como carboxamida, sulfonamida o fosforamida, donde cada alquilo C1-C12, alquiloxi C1-C12
8. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7, o una sal del mismo, junto con uno o más vehículos o adyuvantes farmacéuticamente aceptables.
9. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7, o una sal del mismo, para su uso en un método para tratar una infección bacteriana en un mamífero.
FIGURA 1
Conc. Peptoide de Peptoide de Vancomicina Compuesto-
Pocillo Ensayo Ensayo Control Control Negativo
g/ml Compuesto 1 Compuesto 2
125 62, 5 31, 3 15, 6 7, 8 3, 9 1, 9
Tl Tl Tl T2 T2 T2 VC VC VC NC NC NC
Tl Tl Tl T2 T2 T2 VC VC VC NC NC NC
Tl Tl Tl T2 T2 T2 VC VC VC NC NC NC
Tl Tl Tl T2 T2 T2 VC VC VC NC NC NC
Tl Tl Tl T2 T2 T2 VC VC VC NC NC NC
Tl Tl Tl T2 T2 T2 VC VC VC NC NC NC
Tl Tl Tl T2 T2 T2 VC VC VC NC NC NC
Tl Tl Tl T2 T2 T2 VC VC VC NC NC NC
Patentes similares o relacionadas:
Procedimiento de preparación de cápsula dura de hipromelosa por el uso de termogelificación, del 22 de Julio de 2020, de Suheung Co., Ltd: Un procedimiento de preparación de una cápsula dura de hipromelosa usando termogelificación con estabilidad durante almacenamiento, resistencia de película, distribución […]
Composición para el control de parásitos animales y método para el control de parásitos animales, del 15 de Julio de 2020, de MITSUI CHEMICALS AGRO, INC: Una composición para su uso en un método de tratamiento, en donde el método es para exterminar parásitos animales, comprendiendo la composición, […]
Uso de principios activos refrescantes fisiológicos y agentes que contienen tales principios activos, del 17 de Junio de 2020, de Symrise AG: Procedimiento no terapéutico para la modulación in-vitro del receptor de mentol frío TRPM8, en el que se lleva a contacto el receptor con al menos un modulador, que se selecciona […]
Gránulos de dispersión rápida, comprimidos de desintegración oral y métodos, del 3 de Junio de 2020, de Adare Pharmaceuticals, Inc: Microgránulos de dispersión rápida, farmacéuticamente aceptables, que tienen una mediana del tamaño de partícula en el rango de 100 μm a 300 […]
Composición anestésica de uso tópico en forma de nanopartículas, del 13 de Mayo de 2020, de Biolab Sanus Farmacêutica Ltda: Una composición anestésica en forma de hidrogel de aplicación tópica sobre la piel o las mucosas, que lleva: (i) una suspensión de nanocápsulas […]
Compuestos de sulfonamida inversa a base de sulfuro, alquilo y piridilo para el tratamiento del VHB, del 13 de Mayo de 2020, de Novira Therapeutics Inc: compuesto de Fórmula IIIa: **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo; en la que cada R1 se selecciona independientemente de H, halo […]
Partículas de inhalación que comprenden una combinación de un anticolinérgico, un corticoesteroide y un beta-adrenérgico, del 22 de Abril de 2020, de CHIESI FARMACEUTICI S.P.A.: Micropartículas multicomponentes para uso en una formulación para inhalación, cada micropartícula comprende una combinación de dipropionato de […]
Formulación que comprende glicopirrolato, método y aparato, del 25 de Marzo de 2020, de VECTURA LIMITED: Un método de preparación de una formulación en polvo seco, comprendiendo el método la molienda conjunta por chorro de glicopirrolato sin micronizar […]