DISPERSIONES SOLIDAS COMPRENDIENDO TACROLIMUS.
Composición farmacéutica comprendiendo una solución sólida de tacrolimus disuelta en un vehículo hidrofílico o miscible en agua,
en donde el vehículo es una mezcla de polietilenglicol y poloxámero en una proporción de entre 1:3 y 10: 1, en donde el punto de fusión del vehículo es al menos 20ºC y en donde el tacrolimus está presente en él en una concentración de entre aproximadamente 0.01% p/p y aproximadamente 15% p/p para formar una solución sólida a temperatura ambiente
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/DK2004/000574.
Solicitante: LIFECYCLE PHARMA A/S.
Nacionalidad solicitante: Dinamarca.
Dirección: KOGLE ALLE 4 2970 HORSHOLM DINAMARCA.
Inventor/es: HOLM,PER, NORLING, TOMAS.
Fecha de Publicación: .
Fecha Solicitud PCT: 30 de Agosto de 2004.
Fecha Concesión Europea: 7 de Julio de 2010.
Clasificación PCT:
- A61K31/436 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › conteniendo el sistema heterocíclico un ciclo de seis eslabones teniendo el oxígeno como heteroátomo del ciclo, p. ej. rapamicina.
- A61K9/14 A61K […] › A61K 9/00 Preparaciones medicinales caracterizadas por un aspecto particular. › en estado especial, p. ej. polvos (microcápsulas A61K 9/50).
- A61K9/16 A61K 9/00 […] › Aglomerados; Granulados; Microbolitas.
- A61P37/06 A61 […] › A61P ACTIVIDAD TERAPEUTICA ESPECIFICA DE COMPUESTOS QUIMICOS O DE PREPARACIONES MEDICINALES. › A61P 37/00 Medicamentos para el tratamiento de problemas inmunológicos o alérgicos. › Inmunosupresores, p. ej. medicamentos para el tratamiento del rechazo en injertos.
Clasificación antigua:
- A61K31/436 A61K 31/00 […] › conteniendo el sistema heterocíclico un ciclo de seis eslabones teniendo el oxígeno como heteroátomo del ciclo, p. ej. rapamicina.
- A61K9/14 A61K 9/00 […] › en estado especial, p. ej. polvos (microcápsulas A61K 9/50).
- A61K9/16 A61K 9/00 […] › Aglomerados; Granulados; Microbolitas.
- A61P37/06 A61P 37/00 […] › Inmunosupresores, p. ej. medicamentos para el tratamiento del rechazo en injertos.
Países PCT: Austria, Bélgica, Suiza, Alemania, Dinamarca, España, Francia, Reino Unido, Grecia, Italia, Liechtensein, Luxemburgo, Países Bajos, Suecia, Mónaco, Portugal, Irlanda, Eslovenia, Finlandia, Rumania, Chipre.
Fragmento de la descripción:
Dispersiones sólidas comprendiendo Tacrolimus.
La presente invención se refiere a una solución sólida comprendiendo tacrolimus que tiene una realzada biodisponibilidad, más específicamente una solución sólida de tacrolimus en un vehículo hidrofílico; una composición farmacéutica comprendiendo la solución sólida, y formas de dosificación comprendiendo la solución sólida.
Antecedentes de la invención
El tacrolimus, conocido también como FK-506 o FR-900506, tiene la estructura tricíclica química que se muestra a continuación:
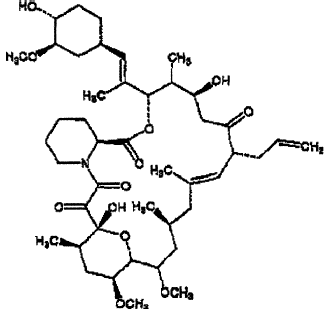
que corresponde a C44 H69 NO12. El tacrolimus aparece en forma de cristales blancos o polvo cristalino. Es prácticamente insoluble en agua, libremente soluble en etanol y muy soluble en metanol y cloroformo.
La preparación de tacrolimus se describe en EP-A-0 184 162 y los análogos de tacrolimus se exponen p. ej. en EP-A-0 444 659 y US 6,387,918.
El tacrolimus es un compuesto macrólido con útil actividad inmunosupresora, actividad antimicrobiana y otras actividades farmacológicas y es de valor para el tratamiento o prevención de reacciones de rechazo por el trasplante de órganos o tejidos, enfermedades de injerto contra huésped, enfermedades autoinmunes y enfermedades infecciosas. El tacrolimus prolonga la supervivencia del huésped y del injerto trasplantado en modelos animales de trasplante de hígado, riñón, corazón, médula ósea e intestino delgado y páncreas, pulmón y tráquea, piel, córnea y extremidad.
En animales, el tacrolimus ha demostrado suprimir alguna inmunidad humoral y, a una mayor extensión, las reacciones mediadas por célula tal como el rechazo al aloinjerto, hipersensibilidad de tipo retardado, artritis inducida por colágeno, encefalomielitis alérgica experimental y la enfermedad del injerto contra huésped.
El tacrolimus inhibe la activación del linfocito T, aunque el mecanismo exacto de acción es desconocido. Evidencia experimental sugiere que el tacrolimus se une a una proteína intracelular, FKBP-12. Un complejo de tacrolimus -FKBP-12, calcio, calmodulina, y calcineurina es formado entonces y la actividad fosfatasa de la calcineurina es inhibida. Este efecto puede impedir la defosforilación y translocación del factor nuclear de las células T activadas, un componente nuclear que se cree que inicia la transcripción genética para la formación de linfoquinas. El resultado neto es la inhibición de la activación de linfocitos T, es decir la inmunosupresión.
El tacrolimus es extensivamente metabolizado por la isoenzima CYP3A4 en la pared del intestino e hígado. Por lo tanto, los fármacos que afectan esta isoenzima pueden influir en la absorción y la posterior eliminación de los tacrolimus sistémicamente absorbidos. Los inhibidores de CYP3A4 pueden aumentar los niveles de tacrolimus, mientras los inductores de CYP3A4 pueden aumentar el metabolismo de tacrolimus y reducir los niveles de tacrolimus. Por consiguiente, el tacrolimus puede ser administrado junto con uno o más inhibidores CYP3A4 a fin de mejorar la biodisponibilidad global.
Normalmente el tacrolimus es administrado por vía oral y por lo tanto es absorbido del tracto gastrointestinal. Se ha observado que la absorción es influenciada negativamente por la ingestión simultánea de alimentos. Así, la velocidad y extensión de la absorción de tacrolimus fueron mayores en condiciones de ayuno.
En general, se conoce que la absorción y biodisponibilidad de una sustancia terapéuticamente activa puede ser afectada por una variedad de factores cuando es administrada por vía oral. Tales factores incluyen la presencia de alimentos en el tracto gastrointestinal y, en general, el tiempo de residencia gástrica de una sustancia farmacológica es significativamente más larga en presencia de alimento que en el estado de ayuno. Si la biodisponibilidad de una sustancia farmacológica es afectada más allá de un cierto punto debido a la presencia de alimentos en el tracto gastrointestinal, se dice que la sustancia farmacológica, exhibe un efecto de alimento. Los efectos de los alimentos son importantes porque la absorción, y por lo tanto, los niveles de plasma pasan a ser altamente variables dependiendo de la ingestión de alimentos. La absorción en el flujo sanguíneo puede ser afectada negativamente, hasta el punto que el paciente se arriesga a la absorción insuficiente para remediar la condición para la que el medicamento se administró. En cambio, las concentraciones de altos valores máximos en condiciones de ayuno, ocasionalmente bien pueden inducir efectos secundarios importantes, de origen nefro- o neuro-tóxico, al igual que efectos secundarios GI y otros.
La absorción del tacrolimus del tracto gastrointestinal después de la administración oral es rápida con un tiempo medio de tiempo-a- concentración máxima (tmax) de aproximadamente 1-2 horas después de la administración a sujetos saludables o pacientes trasplantados de riñón o de hígado, pero incompleto y variable. La biodisponibilidad es generalmente tan baja como, aproximadamente como máximo un 20% después de la administración oral.
Los efectos secundarios frecuentemente observados son vómitos y náuseas pero los efectos secundarios como temblor, dolor de cabeza, hipertensión, disfunción renal, hipercalemia, hipomagnesemia, hiperglicemia, insomnio, diarrea, estreñimiento, dolor abdominal, nefrotoxicidad y neurotoxicidad también son observados.
Para la administración oral, el tacrolimus es actualmente formulado y comercializado como cápsulas de gelatina blanda comprendiendo el equivalente de 0.5, 1 o 5 mg de tacrolimus anhidro y comercializadas bajo el nombre comercial de Prograf® y Protropic®. La recomendada dosis oral inicial es de aproximadamente 0.1 a 0.2 mg/kg/día en pacientes. La dosis pretende una determinada concentración mínima de plasma de aproximadamente 5 a aproximadamente 20 ng/ml. Prograf® es indicado para la profilaxis del rechazo de órganos en pacientes recibiendo trasplantes alogénicos de hígado o riñón.
D1 expone una formulación obtenible pulverizando una solución de tacrolimus, éster de colesterol PEG-24, monoglicéridos y ácido desoxicólico en solvente orgánico sobre semillas "Nonpareil" y se refiere además al realce de la biodisponibilidad oral.
D2 expone formulaciones de liberación sostenida obtenibles disolviendo tacrolimus en monoestearato de glicerol fundido o éster del ácido trigraso de tetraglicerina y mezclando con HPM o lactosa.
D3 expone cápsulas comprendiendo una dispersión sólida de 20% de tacrolimus en HPMC preparadas por un método con solvente.
D4 expone un método de aglomeración controlada para mejorar la biodisponibilidad de compuestos poco solubles en soluciones o dispersiones sólidas.
Sigue habiendo una necesidad de nuevas composiciones farmacéuticas y/o formas de dosificación comprendiendo tacrolimus exhibiendo una realzada biodisponibilidad. Una aumentada biodisponibilidad puede permitir una reducción en las unidades de dosificación tomadas por un paciente, p. ej. bajando a una única dosis diaria, y también puede reducir o anular la necesidad de ingerir alimentos simultáneamente con la forma de dosificación, así permitiendo más libertad a los pacientes sobre cuándo se ingiere el medicamento. Además, se contempla que las fluctuaciones en la concentración de plasma contra el perfil de tiempo pueden ser reducidas significativamente. Además, la realzada biodisponibilidad también puede resultar en un perfil de liberación más reproducible (es decir, menos variable en comparación con aquel del Prograf®).
Breve resumen de la invención
Los inventores han encontrado ahora que la biodisponibilidad de tacrolimus puede ser significativamente realzada disolviendo tacrolimus en un vehículo hidrofílico o miscible en agua en una cantidad que es eficiente para uso en la preparación de una forma útil de dosificación de fármacos. El tacrolimus se conoce por tener una solubilidad muy baja en agua, pero esta invención proporciona composiciones y formulaciones farmacéuticas que exhiben perfiles de liberación in vitro muy rápidos, es decir composiciones de liberación inmediata que son contempladas habiendo aumentado significativamente la biodisponibilidad in vivo en pacientes que lo necesitan.
Por consiguiente, en un primer...
Reivindicaciones:
1. Composición farmacéutica comprendiendo una solución sólida de tacrolimus disuelta en un vehículo hidrofílico o miscible en agua, en donde el vehículo es una mezcla de polietilenglicol y poloxámero en una proporción de entre 1:3 y 10: 1, en donde el punto de fusión del vehículo es al menos 20ºC y en donde el tacrolimus está presente en él en una concentración de entre aproximadamente 0.01% p/p y aproximadamente 15% p/p para formar una solución sólida a temperatura ambiente.
2. Composición según la reivindicación 1, en donde la concentración de tacrolimus en polietilenglicol y poloxámero es como mucho 10% p/p.
3. Composición según la reivindicación 1, en donde la concentración de tacrolimus en polietilenglicol y poloxámero es al menos aproximadamente 0.05% p/p.
4. Composición farmacéutica según la reivindicación 1, en donde al menos 50% p/p de tacrolimus es liberado dentro de aproximadamente 30 minutos, cuando es probado en cualquier prueba de disolución según USP usando un medio de disolución acuoso.
5. Composición farmacéutica según la reivindicación 1, en donde al menos 75% p/p de tacrolimus es liberado dentro de aproximadamente 40 minutos, cuando es probado en cualquier prueba de disolución según USP usando un medio de disolución acuoso.
6. Composición farmacéutica según la reivindicación 1, en donde al menos 90% p/p de tacrolimus es liberado dentro de aproximadamente 60 minutos, cuando es probado en cualquier prueba de disolución según USP usando un medio de disolución acuoso.
7. Composición farmacéutica según la reivindicación 1, en donde el polietilenglicol tiene un peso molecular medio de al menos 1500.
8. Composición farmacéutica según la reivindicación 1, en donde el polietilenglicol y el poloxámero está presente en una proporción de entre 1:1 y 5:1, preferiblemente entre 3:2 y 4:1, más preferiblemente entre 2:1 y 3:1, en particular aproximadamente 7:3.
9. Composición farmacéutica según la reivindicación 1, en donde el poloxámero es el poloxámero 188.
10. Composición farmacéutica según la reivindicación 1, en donde el polietilenglicol tiene un peso molecular medio de aproximadamente 6000 (PEG6000).
11. Composición farmacéutica según cualquiera reivindicación 1 a 10 comprendiendo además uno o más excipientes farmacéuticamente aceptables.
12. Composición farmacéutica según la reivindicación 11, en donde los excipientes farmacéuticamente aceptables son seleccionados del grupo consistiendo en cargas, desintegrantes, ligantes y lubricantes.
13. Composición farmacéutica según la reivindicación 11 en forma particulada, por ejemplo en forma de polvo.
14. Composición farmacéutica según la reivindicación 13, en donde las partículas tienen un diámetro de peso geométrico medio dgw desde aproximadamente 10 μm hasta aproximadamente 2000 μm, preferiblemente desde aproximadamente 20 μm hasta aproximadamente 2000 μm, especialmente desde aproximadamente 50 μm hasta aproximadamente 300 μm.
15. Composición farmacéutica según la reivindicación 13, en donde las partículas tienen un diámetro de peso geométrico medio dgw desde aproximadamente 50 μm hasta aproximadamente 300 μm.
16. Forma de dosificación comprendiendo la composición farmacéutica según la reivindicación 11, que es una forma de dosificación oral sólida.
17. Forma de dosificación según la reivindicación 16, que es una forma de dosificación unitaria.
18. Forma de dosificación según la reivindicación 16, que comprende además un aditivo farmacéuticamente aceptable seleccionado del grupo consistiendo en agentes aromatizantes, agentes colorantes, agentes de enmascarado del sabor, agentes ajustadores de pH, agentes tampón, conservantes, agentes estabilizantes, antioxidantes, agentes humectantes, agentes ajustadores de la humedad, agentes surfactivos, agentes de suspensión, agentes realzadores de la absorción y agentes modificadores de la liberación.
19. Forma de dosificación según la reivindicación 16, en donde al menos un excipiente farmacéuticamente aceptable es seleccionado del grupo consistiendo en ácido de sílice o un derivado o sal del mismo incluyendo silicatos, dióxido de silicio y polímeros de los mismos; aluminosilicato de magnesio y/o aluminometasilicato de magnesio, bentonita, caolín, trisilicato de magnesio, montmorilonita y/o saponita.
20. Forma de dosificación según la reivindicación 16, en donde al menos un excipiente farmacéuticamente aceptable es un ácido de sílice o un derivado o sal del mismo.
21. Forma de dosificación según la reivindicación 16, en donde al menos un excipiente farmacéuticamente aceptable es dióxido de silicio o un polímero del mismo.
22. Forma de dosificación según la reivindicación 18 comprendiendo uno o más agentes modificadores de liberación seleccionados del grupo consistiendo en polímeros miscibles en agua, polímeros, aceites y materiales oleaginosos insolubles en agua.
23. Forma de dosificación según la reivindicación 22, en donde el polímero insoluble en agua es seleccionado del grupo consistiendo en etilcelulosa, acetato de celulosa, nitrato de celulosa, y sus mezclas.
24. Forma de dosificación según la reivindicación 22, en donde el aceite o material oleaginoso es seleccionado del grupo consistiendo en aceites hidrofílicos y hidrofóbicos o materiales oleaginosos.
25. Forma de dosificación según la reivindicación 22, en donde el aceite o material oleaginoso es hidrofílico y seleccionado del grupo consistiendo en glicoles de poliéter tales como polipropilenglicoles; polioxietilenos; polioxipropilenos; poloxámeros; glicéridos poliglicolizados y sus mezclas.
26. Forma de dosificación según la reivindicación 22, en donde el aceite o material oleaginoso es hidrofóbico y seleccionado del grupo consistiendo en hidrocarburos saturados de cadena recta, ésteres de sorbitán, parafinas; grasas y aceites tal como la mantequilla de cacao, sebo bovino, manteca de cerdo, ésteres de polietilenglicol; ácido graso superior tal como ácido esteárico, ácido mirístico, ácido palmítico, alcoholes superiores tal como cetanol, alcohol de estearilo, ceras de punto de fusión bajo tal como monoestearato de glicerilo, monooleato de glicerilo, sebo hidrogenado, alcohol de miristilo, alcohol de estearilo, monoglicéridos sustituidos y/o no sustituidos, diglicéridos sustituidos y/o no sustituidos, triglicéridos sustituidos y/o no sustituidos, cera de abejas amarilla, cera de abejas blanca, cera carnauba, cera de ricino, cera de Japón, monoglicéridos de acetilato; polímeros NVP, polímeros PVP, polímeros acrílicos, y sus mezclas.
27. Forma de dosificación según la reivindicación 26, en donde el aceite o material oleaginoso hidrofóbico tiene un punto de fusión de al menos aproximadamente 20ºC.
28. Forma de dosificación según la reivindicación 22, en donde el polímero miscible en agua es un derivado de celulosa seleccionado del grupo consistiendo en hidroxipropilmetilcelulosa (HPMC), hidroxipropilcelulosa (HPC), metilcelulosa, carboximetilcelulosa sódica, hidroxietil celulosa, poloxámeros, estearatos de polioxietileno, poli-
29. Forma de dosificación según la reivindicación 22, que es entero-recubierta usando un polímero miscible en agua que tiene una solubilidad en agua dependiente del pH.
30. Forma de dosificación según la reivindicación 29, en donde el polímero miscible en agua es seleccionado del grupo consistiendo en poliacrilamidas; derivados de ftalato tales como ftalatos ácidos de carbohidratos incluyendo ftalato acetato de amilosa, ftalato acetato de celulosa, tereftalato acetato de celulosa, isoftalato acetato de celulosa, otros ftalatos de éster de celulosa, ftalatos de éter de celulosa, ftalato de hidroxipropilcelulosa, ftalato acetato de hidroxipropilcelulosa, ftalato hidroxipropiletilcelulosa, ftalato de hidroxipropilmetilcelulosa (HMPCP), ftalato de metilcelulosa, ftalato acetato de metilcelulosa, ftalato acetato de polivinilo, ftalato acetato de polivinilo de hidrógeno, ftalato acetato de celulosa de sodio, ftalato de ácido de almidón; ftalatos de otros compuestos incluyendo ftalato acetato de polivinilo (PVAP); otros derivados de celulosa incluyendo succinato acetato de hidroxipropil metilcelulosa (HPMCAS), carboximetilcelulosa, trimelitato acetato de celulosa; alginatos; carbómeros; derivados de ácido poliacrílico tal como copolímeros de ácido acrílico y de éster acrílico, ácido polimetacrílico y ésteres de los mismos, copolímeros de ácido poli acrílico metacrílico, copolímeros de ácido metacrílico; copolímero de estireno-dibutil ftalato de ácido maléico, copolímero de estireno-ftalato polivinilacetato de ácido maléico, copolímeros de estireno y de ácido maleico; goma laca, glicolato de almidón; polacrilina; copolímeros de acetato de vinilo y de ácido crotónico y sus mezclas.
31. Forma de dosificación según la reivindicación 29, que al ser administrada por vía oral a un mamífero que lo necesita libera como mucho aproximadamente 10% p/p, preferiblemente como mucho aproximadamente 7.5% p/p, más preferiblemente como mucho aproximadamente 5% p/p, especialmente como mucho aproximadamente 2% p/p de la cantidad total de ingrediente activo dentro de las primeras 3 horas, preferiblemente dentro de 2 horas, más preferiblemente dentro de 1 hora, en particular dentro de aproximadamente 30 minutos después de la administración.
32. Forma de dosificación sólida según la reivindicación 16, en donde la forma de dosificación sólida al ser administrada oralmente a un mamífero que lo necesita, libera al menos 50% p/p del ingrediente activo dentro de 24 horas, preferiblemente dentro de 20 horas, más preferiblemente dentro de 18 horas, especialmente dentro de 15 horas, en particular dentro de 12 horas.
33. Uso de la composición según la reivindicación 1 para la preparación de una forma de dosificación oral sólida tal como pastillas, cápsulas o sobres.
34. Uso de la composición según la reivindicación 1 para la preparación de gránulos, granulados, microesferas o nanopartículas.
35. Uso de la composición según la reivindicación 1 para la preparación de una forma de dosificación sólida de liberación inmediata.
36. Uso de la composición según la reivindicación 1 para la preparación de una forma de dosificación sólida de liberación retardada.
37. Uso de la composición según la reivindicación 1 para la preparación de una forma de dosificación tópica.
38. Forma de dosificación según la reivindicación 16 para el uso en el tratamiento de condiciones que responden al tratamiento de tacrolimus.
39. Forma de dosificación según la reivindicación 38 para el uso en el tratamiento de reacciones de rechazo por el trasplante de órganos o tejidos o el tratamiento de enfermedad autoinmune.
40. Forma de dosificación según la reivindicación 16 para uso en el tratamiento de un paciente que lo necesita, con una dosificación una vez al día de tacrolimus de 0.02 mg/kg/día a 0.15 mg/kg/día.
41. Método para la preparación de la composición según la reivindicación 1, el método comprendiendo la fase de disolver tacrolimus en polietilenglicol y poloxámero para obtener una solución sólida a temperatura ambiente.
Patentes similares o relacionadas:
Inmunomoduladores, del 29 de Julio de 2020, de BRISTOL-MYERS SQUIBB COMPANY: Un compuesto de la fórmula (I) **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en donde: A se selecciona de **(Ver fórmula)** en donde: […]
 CD52 soluble para su uso en el tratamiento o la prevención de la esclerosis múltiple o de la artritis reumatoide, del 29 de Julio de 2020, de THE WALTER AND ELIZA HALL INSTITUTE OF MEDICAL RESEARCH: Uno cualquiera o más de:
i) glucoproteína CD52 soluble, ii) una proteína de fusión que comprende la glucoproteína CD52 soluble como una primera proteína, y una segunda proteína;
[…]
CD52 soluble para su uso en el tratamiento o la prevención de la esclerosis múltiple o de la artritis reumatoide, del 29 de Julio de 2020, de THE WALTER AND ELIZA HALL INSTITUTE OF MEDICAL RESEARCH: Uno cualquiera o más de:
i) glucoproteína CD52 soluble, ii) una proteína de fusión que comprende la glucoproteína CD52 soluble como una primera proteína, y una segunda proteína;
[…]
Nuevos moduladores de receptores de fosfato de esfingosina, del 15 de Julio de 2020, de THE SCRIPPS RESEARCH INSTITUTE: Una composicion farmaceutica que comprende un compuesto de formula 265: **(Ver fórmula)** o una sal, estereoisomero, hidrato o solvato farmaceuticamente aceptable […]
Macrocíclico y composición que comprende el mismo, del 8 de Julio de 2020, de Shenzhen TargetRx, Inc: Un compuesto macrocíclico sustituido, que es un compuesto macrocíclico representado por la fórmula (I), o una forma cristalina, una sal farmacéuticamente […]
Compuestos de diaminopirimidilo sustituidos, composiciones de los mismos y procedimientos de tratamiento con ellos, del 17 de Junio de 2020, de SIGNAL PHARMACEUTICALS LLC: Un compuesto de fórmula (I): **(Ver fórmula)** o una sal, un tautómero, un isotopólogo o un estereoisómero farmacéuticamente aceptable […]
Anticuerpos que se unen específicamente a TL1A, del 17 de Junio de 2020, de CEPHALON, INC.: Un anticuerpo recombinante que se une específicamente al ligando 1A de tipo TNF (TL1A) y comprende una región variable de cadena pesada que comprende la secuencia […]
Régimen de dosificación para un agonista del receptor S1P, del 3 de Junio de 2020, de NOVARTIS AG: Uso de un modulador o agonista del receptor S1P en la fabricación de un medicamento para el tratamiento de una enfermedad autoinmune, mediante […]
Anticuerpos anti-IGE humanizados que entrecruzan CD23 en linfocitos B pero no sensibilizan los mastocitos, del 13 de Mayo de 2020, de ACADEMIA SINICA: Un anticuerpo humanizado que tiene una actividad de unión a IgE humana libre, IgE unidas a la membrana en linfocitos B e IgE unida a CD23, pero que es incapaz de unirse a IgE […]