Agentes antibacterianos novedosos.
Un compuesto de formula**Fórmula**
o una sal, hidrato o solvato farmaceuticamente aceptable, donde A es CH2 y B es (CH2)3.
Tipo: Patente Europea. Resumen de patente/invención. Número de Solicitud: E13003970.
Solicitante: MERCK SHARP & DOHME CORP.
Nacionalidad solicitante: Estados Unidos de América.
Dirección: 126 EAST LINCOLN AVENUE RAHWAY, NJ 07065 ESTADOS UNIDOS DE AMERICA.
Inventor/es: HWANG, CHAN, KOU, SHUE, YOUE-KONG, ICHIKAWA, YOSHITAKA, CHIU,YU-HUNG, RABUKA,DAVID, YAO,SULAN, SUCHECK,STEVE, MARBY,KENNETH, LIANG,CHANG-HSING, DUFFIELD,JONATHAN, ROMERO,ALEX.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A61K31/33 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › Compuestos heterocíclicos.
- A61K31/335 A61K 31/00 […] › que tienen el oxígeno como único heteroátomo de un ciclo, p. ej. fungicromina.
- A61K31/555 A61K 31/00 […] › que contienen metales pesados, p. ej. hemina, hematina, melarsoprol.
- A61K31/70 A61K 31/00 […] › Hidratos de carbono; Azúcares; Sus derivados (sorbitol A61K 31/047).
- C07D225/00 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07D COMPUESTOS HETEROCICLICOS (Compuestos macromoleculares C08). › Compuestos heterocíclicos que contienen ciclos de más de siete miembros, que tienen un átomo de nitrógeno como único heteroátomo del ciclo.
- C07D267/02 C07D […] › C07D 267/00 Compuestos heterocíclicos que contienen ciclos de más de seis miembros que tienen un átomo de nitrógeno y un átomo de oxígeno como únicos heteroátomos del ciclo. › Ciclos de siete miembros.
- C07D281/02 C07D […] › C07D 281/00 Compuestos heterocíclicos que contienen ciclos de más de seis miembros que tienen un átomo de nitrógeno y un átomo de azufre como únicos heteroátomos del ciclo. › Ciclos de siete miembros.
- C07D295/00 C07D […] › Compuestos heterocíclicos que contienen ciclos polimetileno-imina de al menos cinco miembros, ciclos aza-3 biciclo [3.2.2] nonano, piperazina, morfolina o tiomorfolina, que tienen solamente átomos de hidrógeno unidos directamente a los átomos de carbono del ciclo.
- C07H17/08 C07 […] › C07H AZUCARES; SUS DERIVADOS; NUCLEOSIDOS; NUCLEOTIDOS; ACIDOS NUCLEICOS (derivados de ácidos aldónicos o sacáricos C07C, C07D; ácidos aldónicos, ácidos sacáricos C07C 59/105, C07C 59/285; cianohidrinas C07C 255/16; glicales C07D; compuestos de constitución indeterminada C07G; polisacáridos, sus derivados C08B; ADN o ARN concerniente a la ingeniería genética, vectores, p. ej. plásmidos o su aislamiento, preparación o purificación C12N 15/00; industria del azúcar C13). › C07H 17/00 Compuestos que contienen radicales heterocíclicos unidos directamente a los heteroátomos de los radicales sacárido. › Heterociclos que contienen ocho o más miembros cíclicos, p. ej. eritromicinas.
PDF original: ES-2552682_T3.pdf
Patentes similares o relacionadas:
Polimorfos de 20,23-piperidinil-5-O-micaminosiltilonolida, del 20 de Mayo de 2020, de INTERVET INTERNATIONAL B.V: Una forma cristalina de 20,23-dipiperidinil-5-O-micaminosil-tilonolida que tiene al menos una de las siguientes características: un espectro FT-Raman que comprende […]
Derivados de azitromicina con propiedades de potenciación de la barrera epitelial, del 13 de Mayo de 2020, de EpiEndo Pharmaceuticals ehf: Un compuesto de acuerdo con la fórmula (I) **(Ver fórmula)** En donde R1 es OH; R2 es de acuerdo con la fórmula (III) **(Ver […]
Derivados de avermectina B1 que tienen un sustituyente alcoximetilo en la posición 4'''' o 4'', del 17 de Febrero de 2020, de Boehringer Ingelheim Animal Health USA Inc: Compuesto de fórmula**Fórmula** en la que n es 0 o 1; A-B es -CH=CH; R1 es sec-butilo o isopropilo; R2 es CH2CH3, p-ClC6H4, CH2(CH2)2N3 […]
Levoisovalerilespiramicina III y preparaciones, métodos de preparación y utilizaciones de la misma, del 1 de Enero de 2020, de Shenyang Fuyang Pharmaceutical Technology Co., Ltd: Compuesto cristalino de levoisovalerilespiramicina III, en el que, la fórmula estructural química de la levoisovalerilespiramicina III se representa como la fórmula […]
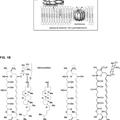 Derivado de anfotericina b con toxicidad reducida, del 25 de Septiembre de 2019, de THE BOARD OF TRUSTEES OF THE UNIVERSITY OF ILLINOIS: C2'epiAmB, representado por**Fórmula**
o una sal farmacéuticamente aceptable del mismo.
Derivado de anfotericina b con toxicidad reducida, del 25 de Septiembre de 2019, de THE BOARD OF TRUSTEES OF THE UNIVERSITY OF ILLINOIS: C2'epiAmB, representado por**Fórmula**
o una sal farmacéuticamente aceptable del mismo.
Forma cristalina de levoisovalerilespiramicina II y preparaciones, procedimientos de preparación y utilizaciones de la misma, del 7 de Agosto de 2019, de Shenyang Fuyang Pharmaceutical Technology Co., Ltd: Compuesto de levoisovalerilespiramicina II, en el que, la fórmula estructural química de la levoisovalerilespiramicina II se representa como la fórmula (II), en condiciones […]
Derivados de la anfotericina B con índice terapéutico mejorado, del 20 de Septiembre de 2018, de The Board of Trustees of the University of Illionis: Un compuesto, o una sal farmacéuticamente aceptable del mismo, el compuesto que se selecciona de**Fórmula**
Procedimientos para tratar enfermedades gastrointestinales, del 11 de Abril de 2018, de Cempra Pharmaceuticals, Inc: Una cantidad terapéuticamente efectiva de un compuesto de fórmula **Fórmula** o una sal farmacéuticamente aceptable del mismo, donde: R10 es hidrógeno […]