Composición farmacéutica y proceso.
Una composición farmacéutica formada por gránulos que contienen la sal clorhidrato del isobutirato de (2R,
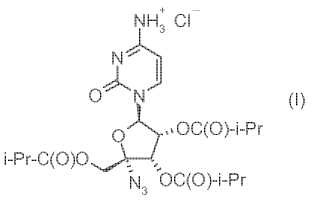
3S, 4R, 5R) -5- (4-amino-2-oxo-2H-pirimidin-1-il) -2-azido-3, 4-bis-isobutiriloxi-tetrahidro-furan-2-ilmetilo (I) y un copolímero de bloques de polietilenglicol (PEG) /polipropilenglicol (PPG), en donde los gránulos se producen mediante una técnica de granulación en húmedo.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/EP2008/053202.
Solicitante: F. HOFFMANN-LA ROCHE AG.
Nacionalidad solicitante: Suiza.
Dirección: GRENZACHERSTRASSE, 124 4070 BASEL SUIZA.
Inventor/es: BIRUDARAJ,KONDAMRAJ, STEFANIDIS,DIMITRIOS, BRANDL,MICHAEL THOMAS, HEGDE,Shridar, SANA,Felix.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A61K9/16 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 9/00 Preparaciones medicinales caracterizadas por un aspecto particular. › Aglomerados; Granulados; Microbolitas.
- A61K9/20 A61K 9/00 […] › Píldoras, pastillas o comprimidos.
PDF original: ES-2377760_T3.pdf
Fragmento de la descripción:
Composición farmacéutica y proceso La invención se refiere a una nueva formulación de tableta comprimida que, como ingrediente activo, tiene el clorhidrato del 2', 3', 5'-triisobutirato de la 4'-azidocitidina y a un proceso para la fabricación de esta formulación. La composición es útil para la terapia del virus de la hepatitis C (VHC) .
Los derivados nucleósidos suelen ser agentes quimioterapéuticos anti-víricos (p.ej. VIH, VHC, herpes simplex, VMC)
y anti-cancerosos potentes. Por desgracia, su utilidad está limitada a menudo por dos factores. En primer lugar, las propiedades farmacocinéticas pobres limiten con frecuencia la absorción del nucleósido en el intestino y la concentración intracelular de los derivados nucleósidos y, en segundo lugar, las propiedades físicas subóptimas restringen las opciones que podrían emplearse para mejorar la entrega del ingrediente activo.
Los profármacos (ver P. Ettmayer y col., J. Med. Chem.47 (10) , 2393-2404, 2004; K. Beaumont y col., Curr. Drug Metab. 4, 461-485, 2003; H. Bundgaard, Design of Prodrugs: Bioreversible derivatives for various functional groups and chemical entities, en: Design of Prodrugs, H. Bundgaard (coord.) , Elsevier Science Publishers, Amersterdam 1985; G.M. Pauletti y col., Adv. Drug Deliv. Rev. 27, 235-256, 1997; R.J. Jones y N. Bischofberger, Antiviral Res. 27, 1-15, 1995 y C.R. Wagner y col., Med. Res. Rev. 20, 417-45, 2000) proporcionan una técnica para mejorar la absorción del fármaco. Los ejemplos típicos de profármacos incluyen los compuestos que tienen grupos protectores biológicamente lábiles unidos a un grupo funcional del compuesto activo. Para el diseño de pronucleótidos se ha recurrido a la alquilación, la acilación u otras modificaciones lipófilas del o de los grupos hidroxi del resto azúcar. Estos pronucleótidos pueden hidrolizarse o desalquilarse “in vivo”, generando de nuevo el compuesto activo. Por desgracia, muchos profármacos que son útiles en otros sentidos presentan una solubilidad acuosa limitada, lo cual plantea retos importantes a la formulación.
Se ha constatado que la 4-amino-1- (5-azido-3, 4-dihidroxi-5-hidroximetil-tetrahidrofuran-2-il) -1H-pirimidin-2-ona (II) es un inhibidor útil de la polimerasa del VHB NS5B; sin embargo, este nucleósido presenta una biodisponibilidad insuficiente. Se ha encontrado que la sal clorhidrato del isobutirato de (2R, 3S, 4R, 5R) -5- (4-amino-2-oxo-2H-pirimidin30 1-il) -2-azido-3, 4-bis-iso-butiriloxi-tetrahidro-furan-2-ilmetilo (I) se absorbe eficazmente y se convierte de nuevo en el compuesto I en el plasma. Lamentablemente, el compuesto I forma un gel cuando se expone al agua, lo cual limita su solubilidad. En este caso, el término “gel” indica una dispersión bifásica que contiene agua y partículas sólidas hidratadas pequeñas del I. Las partículas sólidas dispersadas en agua son difíciles de manipular y procesar para obtener la forma final de dosificación. El gel impide la disolución del ingrediente activo después de la administración. 35 Por tanto, el uso eficaz del compuesto I en la terapia del VHC requiere una formulación que impida la formación del gel durante el procesado, tenga características farmacéuticas apropiadas, se disuelva eficazmente en el intestino y produzca una solución que pueda absorberse en el tracto GI. La terapia antivírica requiere además cantidades relativamente grandes del ingrediente activo para conseguir concentraciones altas del ingrediente activo en suero para evitar la presión de selección de las cepas resistentes. El requisito de que la tableta debe contener niveles elevados del compuesto I limita la cantidad de excipientes, vehículos y diluyentes que pueden incorporarse y aumenta los retos que el formulador tiene afrontar para diseñar una formulación aceptable y eficaz.
La presente invención se refiere a una composición farmacéutica que consta de una tableta comprimida que contiene gránulos la sal clorhidrato del isobutirato de (2R, 3S, 4R, 5R) -5- (4-amino-2-oxo-2H-pirimidin-1-il) -2-azido-3, 4-bis45 iso-butiriloxi-tetrahidrofuran-2-ilmetilo (I; también llamado clorhidrato del 2', 3', 5'-triisobutirato de la 4'-azidocitidina) y un copolímero de bloques de polietilenglicol (PEG) /polipropilenglicol (PPG) .
NH+3 Cl (I)
OC (O) -i-Pr i-Pr-C (O) O OC (O) -i-Pr N3
La presente invención proporciona una composición farmacéutica para la administración oral del clorhidrato del 50 2', 3', 5'-triisobutirato de 4'-azidocitidina que contiene, referidos al peso total de la composición, de 250 mg a 500 mg del clorhidrato del 2', 3', 5'-triisobutirato de 4'-azidocitidina (I) . Este compuesto se describe y reivindica en la patente US-6, 846, 810 publicada en 2 de enero de 2005. Un proceso para la obtención del nucleósido original se describe en T.C. Connolly y col. en la publicación US-20050038240 publicada el 17 de febrero de 2005.
Se ha constatado que el nucleósido triacilado I disminuye la carga vírica en pacientes infectados con el virus de la hepatitis C (VHC) . El virus de la hepatitis C es la causa principal de la enfermedad hepática crónica a nivel mundial (N. Boyer y col., J. Hepatol. 32, 98-112, 2000) . Los pacientes infectados con el VHC corren el riesgo de desarrollar cirrosis de hígado y el posterior carcinoma hepatocelular y por ello el VHC es la principal indicación para el trasplante de hígado. Aunque el compuesto I está disponible en forma cristalina, sus propiedades físicoquímicas dependen del pH. Además, el compuesto forma fácilmente un gel cuando se expone al agua y es difícil de procesar en soluciones acuosas.
Desde hace mucho tiempo, las tabletas son la forma sólida de dosificación preferida. Las tabletas que contienen sustancias farmacológicas pueden fabricarse por técnicas de compresión o de moldeo y pueden contener, o no, diluyentes y/o excipientes adicionales. Normalmente se realiza la compresión de ingredientes granulados solos o en combinación con aglutinantes, desintegrantes, polímeros de liberación controlada, lubricantes o diluyentes. La granulación es un proceso en el que se forman gránulos a partir de sustancia farmacológica en masa, con o sin excipientes, para mejorar las propiedades del fármaco o formulación en masa. Los gránulos son formulaciones que contienen aglomerados sólidos secos de partículas pulverulentas, que son lo suficientemente robustas para soportar el procesado.
Los ingredientes activos granulados se obtienen normalmente por granulación húmeda, granulación en lecho fluidizado o granulación seca. La granulación húmeda se diferencia de la granulación seca porque para el procesado para producir material granulado se emplea un líquido de granulado, por ejemplo agua, líquidos orgánicos o mezclas de los mismos. Las ventajas de la granulación húmeda incluyen la mejora de la cohesión y la compactabilidad de los polvos, el aumento de la densidad, la buena distribución que proporciona un contenido uniforme de fármacos de dosificación baja, micronizados o molidos finamente, la reducción de la contaminación por polvillo y del aire ambiental y la prevención de la segregación de los componentes. Los gránulos de buena calidad tienen normalmente poca cantidad de partículas finas, un tamaño uniforme y permanecen intactos después del secado y de la clasificación. La clasificación puede ir acompañada de tamizado o molienda, por ejemplo. Las tabletas de la invención se fabrican por granulación húmeda, pero los expertos en la materia podrán apreciar que se pueden aplicar otras técnicas de granulación que también están dentro del alcance de la invención.
Se pueden incorporar tensioactivos para mejorar la solubilidad y la dispersabilidad del ingrediente activo, pero los tensioactivos añadidos y otros excipientes tendrán que permitir la producción de una tableta que tenga características aceptables, por ejemplo coherencia y compactabilidad de los polvos, densidad aceptable, dureza, buena distribución que proporcione un contenido uniforme, reducción del polvillo y la contaminación del aire ambiental y prevención de la segregación de los componentes, Por tanto, un objetivo de la invención es una formulación aceptable que pueda facilitar significativamente la tarea del formulador.
Tradicionalmente, la aglomeración en húmedo ha sido una técnica empírica, en la que es difícil prever y explicar el comportamiento observado (S. Iveson y col., Powder Technol. 117, 3-39, 2001) . Un aspecto crítico del desarrollo de una formulación de granulación húmeda satisfactoria consiste en identificar un aglutinante que recubra de... [Seguir leyendo]
Reivindicaciones:
1. Una composición farmacéutica formada por gránulos que contienen la sal clorhidrato del isobutirato de (2R, 3S, 4R, 5R) -5- (4-amino-2-oxo-2H-pirimidin-1-il) -2-azido-3, 4-bis-isobutiriloxi-tetrahidro-furan-2-ilmetilo (I) y un copolímero de bloques de polietilenglicol (PEG) /polipropilenglicol (PPG) , en donde los gránulos se producen mediante una técnica de granulación en húmedo.
2. Una composición según la reivindicación 1 que además contiene por lo menos un diluyente, vehículo y/o excipiente. 10
3. Una composición farmacéutica según la reivindicación 1, en la que dicho copolímero de bloques PEG/PPG es un poloxámero.
4. Una composición farmacéutica según la reivindicación 3 que además contiene por lo menos un diluyente, vehículo 15 y/o excipiente.
5. Una composición farmacéutica según la reivindicación 4, en la que dichos gránulos contienen el compuesto I y poloxámero 188.
6. Una composición farmacéutica según la reivindicación 5, en la que dichos gránulos contienen un 5-20 % (peso/peso) de poloxámero 188.
7. Una composición farmacéutica según la reivindicación 6, en la que dichos gránulos se moldean para obtener una tableta comprimida, comprendiendo dicha tableta además opcionalmente por lo menos un diluyente, un 25 desintegrante, un lubricante y un material de recubrimiento.
8. Una composición farmacéutica según la reivindicación 7, en la que dichos diluyentes son manitol y opcionalmente MCC, dicho desintegrantes es croscarmelosa y dicho lubricante es estearato magnésico.
9. Una composición farmacéutica según la reivindicación 8, en donde dicha tableta comprimida consta de:
ingrediente % de la composición en peso compuesto I poloxámero 188 celulosa microcristalina silicificada manitol croscarmelosa sódica estearato magnésico 50-70 2-8 5-30 2-20 3-20 0, 3-2en donde dicha tableta comprimida está opcionalmente recubierta con Opadr y yellow 03K 12429 y el peso total de dicha tableta comprimida se sitúa entre 100 y 1200 mg.
10. Una composición farmacéutica según la reivindicación 7, en la que dicha tableta comprimida consta de:
ingrediente % de la composición en peso compuesto I poloxámero 188 celulosa microcristalina silicificada manitol croscarmelosa sódica estearato magnésico 55-65 5-7 15-25 3-7 8-12 1, 2-1, 8en donde dicha tableta comprimida está opcionalmente recubierta con Opadr y yellow 03K 12429 y el peso total de dicha tableta comprimida se sitúa entre 100 y 1200 mg.
11. Una composición farmacéutica según la reivindicación 9, en la que el peso total de dicha tableta comprimida es de 500 a 1000 mg.
Patentes similares o relacionadas:
Preparación sólida que contiene colorante, del 29 de Julio de 2020, de DAIICHI SANKYO COMPANY, LIMITED: Preparación farmacéutica sólida que comprende monobencenosulfonato de ácido [(1R,5S,6S)-6-(aminometil)-3- etilbiciclo[3.2.0]hept-3-en-6-il]acético […]
Formulación de vitamina D de liberación modificada estabilizada y método de administración de la misma, del 22 de Julio de 2020, de EirGen Pharma Ltd: Una formulacion oral de liberacion controlada de un compuesto de vitamina D que comprende uno o ambos de 25- hidroxivitamina D2 y 25-hidroxivitamina D3, la formulacion […]
Métodos y composiciones para la administración oral de proteínas, del 22 de Julio de 2020, de Entera Bio Ltd: Una única composición farmacéutica oral que comprende una proteína que tiene un peso molecular de hasta 100.000 Daltons, siendo dicha proteína PTH; […]
Composición farmacéutica que comprende un agente antipsicótico atípico y método para su preparación, del 15 de Julio de 2020, de PHARMATHEN S.A.: Comprimido de liberación controlada de Paliperidona en forma de comprimido de varias capas que comprende: a) un núcleo de matriz que comprende […]
Composiciones y métodos para tratar el virus de la hepatitis C, del 15 de Julio de 2020, de Gilead Pharmasset LLC: Una composición farmacéutica que comprende: a) de aproximadamente el 25% a aproximadamente el 35% p/p de GS-7977 cristalino que tiene la estructura **(Ver […]
Macrogols para aplicación a la mucosa, y sus usos terapéuticos, del 15 de Julio de 2020, de S.I.I.T. S.R.L.-SERVIZIO INTERNAZIONALE IMBALLAGGI TERMOSALDANTI: Composición farmacéutica en forma sólida que comprende, por unidad de dosificación, entre 5 y 400 mg de un PEG con un grado de 3000 o más, para uso en el tratamiento […]
Granulados secos de polvos de sílice mesoporosa, del 1 de Julio de 2020, de FORMAC PHARMACEUTICALS N.V: Un granulado seco que comprende desde el 50% al 100% p/p de sílice mesoporosa ordenada que tiene una organización bidimensional hexagonalmente […]
Composición farmacéutica novedosa, del 1 de Julio de 2020, de NOVARTIS AG: Un comprimido farmaceutico que comprende: a) un farmaco que es el solvato con sulfoxido de dimetilo de N-{3-[3-ciclopropil-5-(2-fluoro-4-5 yodofenilamino)-6,8- dimetil-2,4,7-trioxo-3,4,6,7-tetrahidro-2H-pirido[4,3-d]pirimidin-1-il]fenil}acetamida […]