DERIVADOS DE PIRROLOPIRIDINA COMO INHIBIDORES DE PROTEINA QUINASAS.
Un compuesto seleccionado entre la Fórmula Ia, Ib y Ic:
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/US2006/018868.
Solicitante: IRM LLC
THE SCRIPPS RESEARCH INSTITUTE.
Nacionalidad solicitante: Bermuda.
Dirección: HURST HOLME, 12 TROTT ROAD,HAMILTON, HM 11.
Inventor/es: GRAY,NATHANAEL S, OKRAM,BARUN, REN,PINGDA.
Fecha de Publicación: .
Fecha Concesión Europea: 26 de Mayo de 2010.
Clasificación Internacional de Patentes:
- C07D471/04 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07D COMPUESTOS HETEROCICLICOS (Compuestos macromoleculares C08). › C07D 471/00 Compuestos heterocíclicos que contienen átomos de nitrógeno como únicos heteroátomos del sistema condensado, teniendo al menos un ciclo de seis miembros con un átomo de nitrógeno, no previstos en los grupos C07D 451/00 - C07D 463/00. › Sistemas condensados en orto.
Clasificación PCT:
- A61K31/437 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › conteniendo el sistema heterocíclico un ciclo de cinco eslabones teniendo el nitrógeno como heteroátomo del ciclo, p. ej. indolicina, beta-carbolina.
- A61P35/00 A61 […] › A61P ACTIVIDAD TERAPEUTICA ESPECIFICA DE COMPUESTOS QUIMICOS O DE PREPARACIONES MEDICINALES. › Agentes antineoplásicos.
- C07D471/04 C07D 471/00 […] › Sistemas condensados en orto.
Fragmento de la descripción:
Derivados de pirrolopiridina como inhibidores de proteína quinasas.
Antecedentes de la invención
La invención proporciona una nueva clase de compuestos, composiciones farmacéuticas que comprenden estos compuestos y métodos de uso de estos compuestos para tratar o prevenir enfermedades o trastornos asociados con una actividad quinasa anormal o desregulada, especialmente enfermedades o trastornos que implican la activación anormal de las quinasas CDK, Aurora, Jak2, Rock, CAMKII, FLT3, Tie2, TrkB, FGFR3 y KDR.
Las proteína quinasas representan una gran familia de proteínas que desempeñan una función central en la regulación de una amplia variedad de procesos celulares y que mantienen el control sobre la función celular. Una lista parcial, no limitante, de estas quinasas incluye: tirosina quinasas tales como FLT3, Tie2, TrkB, KDR y el receptor del factor de crecimiento de fibroblastos, FGFR3; y serina/treonina quinasas tales como CDK, Aurora, Jak2, Rock y CAMKII. Se ha observado actividad quinasa aberrante en muchos estados de enfermedad, incluyendo trastornos proliferativos benignos y malignos así como enfermedades que son el resultado de una activación inadecuada de los sistemas inmune y nervioso.
Los nuevos compuestos de esta invención inhiben la actividad de una o más proteína quinasas y, por lo tanto, se espera que sean útiles en el tratamiento de enfermedades asociadas con quinasas.
El documento WO 2004/016610 describe compuestos de la siguiente fórmula general, que se supone que tienen actividad como inhibidores de la quinasa Itk:
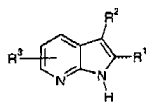
En esta fórmula, el grupo R1 representa un grupo fenilo.
El documento WO 98/47899 describe compuestos de la siguiente fórmula general, que se supone que tienen actividad como inhibidores de la producción de varias citocinas inflamatorias:
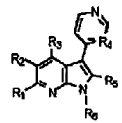
En esta fórmula, el grupo R5 representa un grupo fenilo.
El documento WO 02/04447 describe compuestos de la siguiente fórmula general, que se supone que muestran actividad anti-tumoral:
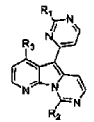
El documento WO 2005/095400 se publicó el 13 de octubre de 2005, después de la fecha de prioridad de la presente solicitud y antes de la fecha de presentación de la presente solicitud. Entró en la Fase Regional Europea, y por lo tanto es de relevancia bajo el Artículo 54(3) EPC para las partes de la presente solicitud otorgadas a su fecha de prioridad. Este documento describe compuestos de la siguiente fórmula general, que se supone que tienen actividad como inhibidores de JAK y otras quinasas:
Descripción resumida de la invención
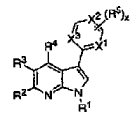
En un aspecto, la presente invención proporciona compuestos seleccionados entre la Fórmula Ia, Ib y Ic:
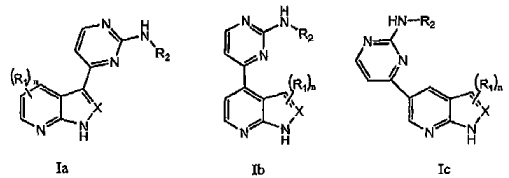
en las que:
n se selecciona entre 0, 1 y 2;
R1 se selecciona entre halo, alquilo C1-6, alcoxi C1-6, alquilo C1-6 halo-sustituido y alcoxi C1-6 halo-sustituido;
R2 se selecciona entre arilo C6-10-alquilo C0-4 y heteroarilo C5-10-alquilo C0-4; donde dicho arilo o heteroarilo de R2 está opcionalmente sustituido con 1-3 radicales seleccionados independientemente entre halo, alquilo C1-6, alcoxi C1-6, alquilo C1-6 halo-sustituido, alcoxi C1-6 halo-sustituido, -(O)0-2R5, -COOR5, -C(O)NR5R6 y
X se selecciona entre CR7 o N; donde R7 se selecciona entre hidrógeno y alquilo C1-6; y las sales farmacéuticamente aceptables, hidratos y solvatos de los mismos; con la condición de que el compuesto no sea

En un segundo aspecto, la presente invención proporciona una composición farmacéutica que contiene una cantidad terapéuticamente eficaz de un compuesto de Fórmula I o una sal farmacéuticamente aceptable, hidrato o solvato del mismo, mezclado con uno o más excipientes adecuados.
En este documento también se describe un procedimiento para tratar una enfermedad en un animal en el que la inhibición de la actividad quinasa, particularmente la actividad de CDK, Aurora, Jak2, Rock, CAMKII, FLT3, Tie2, TrkB, FGFR3 y/o KDR, puede prevenir, inhibir o mejorar la patología y/o sintomatología de la enfermedad, comprendiendo el procedimiento administrar al animal una cantidad terapéuticamente eficaz de un compuesto de Fórmula I o un derivado de N-óxido, isómeros individuales y mezclas de isómeros de los mismos, o una sal farmacéuticamente aceptable de los mismos.
En un tercer aspecto, la presente invención proporciona el uso de un compuesto de Fórmula I en la preparación de un medicamento para tratar una enfermedad en un animal donde la actividad quinasa, particularmente la actividad de CDK, Aurora, Jack2, Rock, CAMKII, FLT3, Tie2, TrkB, FGFR3 y/o KDR, contribuye a la patología y/o sintomatología de la enfermedad.
En un cuarto aspecto, la presente invención proporciona un procedimiento para preparar compuestos de Fórmula I y los derivados de N-óxido, derivados de profármaco, derivados protegidos, isómeros individuales y mezclas de isómeros de los mismos, y las sales farmacéuticamente aceptables de los mismos.
Descripción detallada de la invención
Definiciones
"Alquilo" como un grupo y como un elemento estructural de otros grupos, por ejemplo, alquilo halo-sustituido y alcoxi, puede ser de cadena lineal o ramificada. Alcoxi C1-4 incluye metoxi, etoxi y similares. Alquilo halo-sustituido incluye trifluorometilo, pentafluoroetilo y similares.
"Arilo" se refiere a una unión de anillo aromático monocíclico o bicíclico condensado que contiene de seis a diez átomos de carbono en el anillo. Por ejemplo, arilo puede ser fenilo o naftilo, preferiblemente fenilo. "Arileno" se refiere a un radical divalente obtenido a partir de un grupo arilo.
"Heteroarilo" es como se ha definido para arilo anteriormente donde uno o más de los miembros carbono del anillo indicados pueden reemplazarse por un heteroátomo. Por ejemplo, heteroarilo C5-8 incluye piridilo, indolilo, indazolilo, quinoxalinilo, quinolinilo, benzofuranilo, benzopiranilo, benzotiopiranilo, benzo[1,3]dioxol, imidazolilo, benzo-imidazolilo, pirimidinilo, furanilo, oxazolilo, isoxazolilo, triazolilo, tetrazolilo, pirazolilo, tienilo, etc.
"Cicloalquilo" se refiere a una unión de anillo monocíclico, bicíclico condensado o policíclico puenteado, saturado o parcialmente insaturado, que contiene el número indicado de átomos en el anillo. Por ejemplo, cicloalquilo C3-10 incluye ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, etc.
"Heterocicloalquilo" se refiere a cicloalquilo, como se define en esta solicitud, con la condición de que uno o más de los carbonos indicados del anillo se reemplacen por un resto seleccionado entre
Reivindicaciones:
1. Un compuesto seleccionado entre la Fórmula Ia, Ib y Ic:
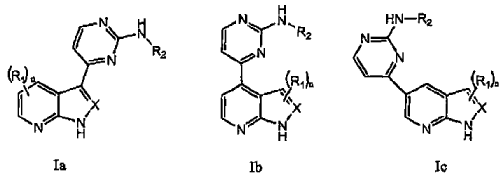
en las que:
con la condición de que el compuesto no sea

2. El compuesto de la reivindicación 1 en el que;
n se selecciona entre 0 y 1;
R1 es alcoxi C1-6;
R2 se selecciona entre arilo C6-10-alquilo C0-4 y heteroarilo C5-10-alquilo C0-4;
donde dicho arilo o heteroarilo de R2 está opcionalmente sustituido con 1-3 radicales seleccionados independientemente entre halo, alquilo C1-6, alcoxi C1-6, alquilo C1-6 halo-sustituido, -S(O)0-2R5, -COOR5, y
3. El compuesto de la reivindicación 2 en el que: R1 es metoxi; R2 se selecciona entre fenilo, bencilo y piridinilo; donde dicho fenilo, bencilo o piridinilo de R2 está opcionalmente sustituido con 1 a 2 radicales seleccionados independientemente entre cloro, bromo, flúor, metilo, trifluorometoxi, trifluorometilo, -COO2R5, -S(O)2R5 y
4. El compuesto de la reivindicación 1 seleccionado entre (3-Cloro-fenil)-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; (4-Fluoro-fenil)-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; [4-(1H-Pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-(4-trifluorometoxi-fenil)-amina; [4-(1H-Pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-(3-trifluorometil-fenil)-amina; (3,4-Difluoro-fenil)-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; [4-(1H-Pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-(4-trifluorometil-fenil)-amina; Éster etílico del ácido 4-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-ilamino]-benzoico; (3-Metoxi-fenil)-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; (3-Fluoro-bencil)-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; (3,5-Dimetoxi-fenil)-(4-(1H-pirrolo[2,3-b]piridin-4-il)pirimidin-2-il]-amina; (2-Metil-piridin-4-il)-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; (2-Cloro-piridin-4-il)-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; (2-Metoxi-piridin-4-il)-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; (4-Metanosulfonil-fenil)-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; Piridin-4-il-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; (4-Metil-pirimidin-2-il)-[4-(1H-pirrolo(2,3-b]piridin-4-il)-pirimidin-2-il]-amina; (3-Bromofenil)-[4-(1-metil-1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; (3-Clorofenil)-[4-(1-metil-1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; [4-(1-Metil-1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-(3-trifluorometil-fenil)-amina; (3-Bromo-4-metil-fenil)-[4-(1-metil-1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; 2-{4-[2-(3-Bromo-fenilamino)-pirimidin-4-il]-pirrolo[2,3-b]piridin-1-il}-etanol; 2-{4-[2-(3-Trifluorometil-fenilamino)pirimidin-4-il]-pirrolo[2,3-b]piridin-1-il}-etanol; 2-{4-[2-(3-Cloro-fenilamino)-pirimidin-4-il]-pirrolo[2,3-b]piridin-1-il}-etanol; 2-{4-[2-(3-Bromo-4-metil-fenilamino)-pirimidin-4-il]-pirrolo[2,3-b]piridin-1-il}-etanol; {4-[1-(2-Amino-etil)-1H-pirrolo[2,3-b]piridin-4-il]-pirimidin-2-il}-(3-bromo-fenil)-amina; {4-[1-(2-Amino-etil)-1H-pirrolo[2,3-b]piridin-4-il]-pirimidin-2-il}-(3-bromo-fenil)-metil-amina; {4-[1-(2-Amino-etil)-1H-pirrolo[2,3-b]piridin-4-il]-pirimidin-2-il}-(4-trifluorometil-fenil)-amina; N-{4-Metil-3-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-ilamino]-fenil}-3-trifluorometil-benzamida; [4-(1H-Pirrolo[2,3-b]piridin-3-il)-pirimidin-2-il]-(4-trifluorometil-fenil)-amina; (3,5-Dimetoxi-fenil)-[4-(1H-pirrolo[2,3-b]piridin-3-il)-pirimidin-2-il]-amina; (3,5-Difluoro-fenil)-[4-(1H-pirrolo[2,3-b]piridin-3-il)-pirimidin-2-il]-amina; (3-Bromofenil)-[4-(1H-pirrolo[2,3-b]piridin-3-il)-pirimidin-2-il]-amina; (3-Metoxi-fenil)-[4-(1H-pirrolo[2,3-b]piridin-3-il)-pirimidin-2-il]-amina; (4-Cloro-fenil)-[4-(1H-pirrolo[2,3-b]piridin-3-il)-pirimidin-2-il]-amina; Éster etílico del ácido 4-[4-(1H-pirrolo[2,3-b]piridin-3-il)-pirimidin-2-ilamino]-benzoico; N-{4-Metil-3-[4-(1H-pirrolo[2,3-b]piridin-3-il)-pirimidin-2-ilamino]-fenil}-3-trifluorometil-benzamida; (3-Clorofenil)-[4-(4-metoxi-1H-pirrolo[2,3-b]piridin-3-il)-pirimidin-2-il]-amina; [4-(4-Metoxi-1H-pirrolo[2,3-b]piridin-3-il)-pirimidin-2-il]-(3-trifluorometil-fenil)-amina; (3-Bromo-fenil)-[4-(4-metoxi-1H-pirrolo[2,3-b]piridin-3-il)-pirimidin-2-il]-amina; N-{4-Metil-3-[4-(1H-pirrolo[2,3-b]piridin-5-il)-pirimidin-2-ilamino]-fenil}-3-trifluorometil-benzamida; N-Etil-4-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-ilamino]-bencenosulfonamida; N-(2-Metoxi-etil)-4-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-ilamino]-bencenosulfonamida; N-(3-Metoxi-propil)-4-[4-(1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-ilamino]-bencenosulfonamida; N-(3-Metoxi-propil)-4-[4-(1H-pirrolo[2,3-b]piridin-3-il)-pirimidin-2-ilamino]-bencenosulfonamida; (3-Bromofenil)-[4-(2-metil-1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; (3-Clorofenil)-[4-(2-metil-1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-amina; [4-(2-Metil-1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-il]-(3-trifluorometil-fenil)-amina; N-(2-Metoxi-etil)-4-[4-(2-metil-1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-ilamino]-bencenosulfonamida; N-(3-Metoxi-propil)-4-[4-(2-metil-1H-pirrolo[2,3-b]piridin-4-il)-pirimidin-2-ilamino]-bencenosulfonamida; (3-Bromo-fenil)-[4-(1H-pirazolo[3,4-b)piridin-4-il)-pirimidin-2-il]-amina; (3-Cloro-fenil)-[4-(1H-pirazolo[3,4-b]piridin-4-il)-pirimidin-2-il]-amina; [4-(1H-Pirazolo[3,4-b]piridin-4-il)-pirimidin-2-il]-(3-trifluorometil-fenil)-amina; (3-Cloro-fenil)-[4-(1H-pirrolo[2,3-b]piridin-5-il)-pirimidin-2-il]-amina; (3-Bromo-fenil)-[4-(1H-pirrolo[2,3-b]piridin-5-il)-pirimidin-2-il]-amina; y [4-(1H-Pirrolo[2,3-b]piridin-5-il)-pirimidin-2-il]-(3-trifluorometilfenil)-amina.
5. Una composición farmacéutica que comprende una cantidad terapéuticamente efectiva de un compuesto de la reivindicación 1 en combinación con un excipiente farmacéuticamente aceptable.
6. Un compuesto de la reivindicación 1 para su uso en el tratamiento de una enfermedad en la que la inhibición de la actividad quinasa puede prevenir, inhibir o mejorar la patología y/o sintomatología de la enfermedad.
7. Un compuesto de acuerdo con la reivindicación 6, en el que la quinasa se selecciona entre CDK, Aurora, Jak 2, Rock, CAMKII, FLT3, Tie2, TrkB, FGFR3 y KDR.
8. El uso de un compuesto de la reivindicación 1 en la preparación de un medicamento para tratar una enfermedad en un animal en el que la actividad quinasa de CDK, Aurora, Jak2, Rock, CAMKII, FLT3, Tie2, TrkB, FGFR3 y KDR contribuye a la patología y/o sintomatología de la enfermedad.
Patentes similares o relacionadas:
Compuestos y procedimientos de uso, del 29 de Julio de 2020, de Medivation Technologies LLC: Un compuesto de fórmula (Aa-1): **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en la que: A representa H, halógeno, amino, […]
Compuestos de heteroaril carboxamida como inhibidores de RIPK2, del 29 de Julio de 2020, de BOEHRINGER INGELHEIM INTERNATIONAL GMBH: Un compuesto de fórmula (I): **(Ver fórmula)** o sus sales farmacéuticamente aceptables, en la que: X es N y Y es CH; o X es CH y Y es N; […]
Compuestos de alquinilbenceno heterocíclicos, y composiciones médicas y usos de los mismos, del 29 de Julio de 2020, de Guangzhou Healthquest Pharma Co., Ltd: Un compuesto de alquinilbenceno heterocíclico que tiene la fórmula (I) y una sal farmacéuticamente aceptable, o estereoisómero del mismo, **(Ver […]
Derivados de piperidina 1,4 sustituidos, del 29 de Julio de 2020, de 89Bio Ltd: Un compuesto de acuerdo con la Fórmula I: **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en donde: A se selecciona de […]
Ureas cíclicas como inhibidores de ROCK, del 22 de Julio de 2020, de BRISTOL-MYERS SQUIBB COMPANY: Un compuesto de acuerdo con la Fórmula (I): **(Ver fórmula)** o un enantiómero, un diastereómero, un estereoisómero, un tautómero, una sal farmacéuticamente aceptable […]
Formas sólidas de un compuesto modulador de quinasas, del 22 de Julio de 2020, de PLEXXIKON, INC: Una forma cristalina del Compuesto I: **(Ver fórmula)** que es la Forma C del Compuesto I caracterizado por un difractograma de rayos […]
Derivado heteroarilo o sal farmacéuticamente aceptable del mismo, método de preparación del mismo y composición farmacéutica para prevenir o tratar enfermedades asociadas con PI3 quinasas, que contiene el mismo como principio activo, del 22 de Julio de 2020, de KOREA RESEARCH INSTITUTE OF CHEMICAL TECHNOLOGY: Un compuesto representado por la fórmula 1, un isómero óptico del mismo o una sal farmacéuticamente aceptable del mismo: **(Ver fórmula)** en la fórmula […]
Procedimiento de preparación de la forma A de grapiprant, del 22 de Julio de 2020, de Aratana Therapeutics Inc: Un procedimiento de preparación de una Forma A cristalina sustancialmente pura de grapiprant, comprendiendo el procedimiento: i. poner en contacto grapiprant a temperatura […]