Proceso para preparar una composición de inmunoglobulina.
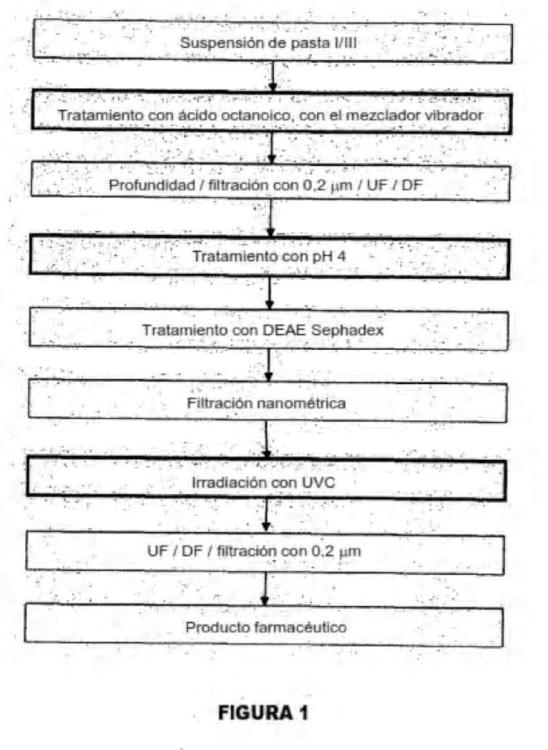
Un proceso para la preparación de una composición de inmunoglobulina que contiene IgM a partir de una fracción de plasma que comprende inmunoglobulinas,
comprendiendo el proceso:
(a) proporcionar una fracción plasmática en forma de solución que contiene las inmunoglobulinas;
(b) mezclar un ácido carboxílico C7 a C9 con la solución y tratar la solución mezclada con un agitador vibrador para precipitar las proteínas contaminantes; y
(c) separar las proteínas precipitadas a partir de la solución para dar como resultado la composición de inmunoglobulina que contiene la IgM.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/EP2011/056486.
Solicitante: BIOTEST AG.
Nacionalidad solicitante: Alemania.
Dirección: LANDSTEINERSTR. 5 63303 DREIEICH ALEMANIA.
Inventor/es: RUDNICK, DIETER, DR., MOLLER, WOLFGANG, DR., MANEG,OLIVER, RODEMER,MICHAEL, DICHTELMUELLER,HERBERT, FLECHSIG,ECKHARD.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A61K39/395 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 39/00 Preparaciones medicinales que contienen antígenos o anticuerpos (materiales para ensayos inmunológicos G01N 33/53). › Anticuerpos (aglutininas A61K 38/36 ); Inmunoglobulinas; Inmunosuero, p. ej. suero antilinfocitario.
- A61K9/08 A61K […] › A61K 9/00 Preparaciones medicinales caracterizadas por un aspecto particular. › Soluciones.
- A61L2/10 A61 […] › A61L PROCEDIMIENTOS O APARATOS PARA ESTERILIZAR MATERIALES U OBJECTOS EN GENERAL; DESINFECCION, ESTERILIZACION O DESODORIZACION DEL AIRE; ASPECTOS QUIMICOS DE VENDAS, APOSITOS, COMPRESAS ABSORBENTES O ARTICULOS QUIRURGICOS; MATERIALES PARA VENDAS, APOSITOS, COMPRESAS ABSORBENTES O ARTICULOS QUIRURGICOS (conservación de cuerpos o desinfección caracterizada por los agentes empleados A01N; conservación, p. ej. esterilización de alimentos o productos alimenticios A23; preparaciones de uso medico, dental o para el aseo A61K). › A61L 2/00 Procedimientos o aparatos para desinfectar o esterilizar materiales u objetos distintos a los productos alimenticios y a las lentes de contacto; Sus accesorios (pulverizadores de desinfectantes A61M; esterilización de envases o del contenido del envase asociado a su contenedor B65B 55/00; tratamiento del agua, agua residual o de alcantarilla C02F; desinfección del papel D21H 21/36; dispositivos de desinfección para retretes E03D; artículos que incluyen accesorios para la desinfección, ver las subclases apropiadas para estos artículos, p. ej. H04R 1/12). › Ultravioleta.
- A61P31/00 A61 […] › A61P ACTIVIDAD TERAPEUTICA ESPECIFICA DE COMPUESTOS QUIMICOS O DE PREPARACIONES MEDICINALES. › Antiinfecciosos, es decir antibióticos, antisépticos, quimioterápicos.
- A61P37/00 A61P […] › Medicamentos para el tratamiento de problemas inmunológicos o alérgicos.
- C07K16/06 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07K PEPTIDOS (péptidos que contienen β -anillos lactamas C07D; ipéptidos cíclicos que no tienen en su molécula ningún otro enlace peptídico más que los que forman su ciclo, p. ej. piperazina diones-2,5, C07D; alcaloides del cornezuelo del centeno de tipo péptido cíclico C07D 519/02; proteínas monocelulares, enzimas C12N; procedimientos de obtención de péptidos por ingeniería genética C12N 15/00). › C07K 16/00 Inmunoglobulinas, p. ej. anticuerpos mono o policlonales. › del suero.
PDF original: ES-2553385_T3.pdf
Patentes similares o relacionadas:
Formulaciones estabilizadas que contienen anticuerpos anti-receptor de interleucina 4 (IL-4R), del 29 de Julio de 2020, de REGENERON PHARMACEUTICALS, INC.: Una jeringuilla precargada que contiene una formulación farmacéutica líquida estable, en la que la formulación farmacéutica líquida comprende: […]
Composición de anticuerpos monoclonales dirigidos contra BDCA-2, del 22 de Julio de 2020, de LABORATOIRE FRANCAIS DU FRACTIONNEMENT ET DES BIOTECHNOLOGIES: Composición de anticuerpos monoclonales dirigidos contra la proteína BDCA-2, presentando dichos anticuerpos un porcentaje de fucosilación inferior al 60% […]
Anticuerpos anti-PD-L1 y usos de los mismos, del 22 de Julio de 2020, de MERCK PATENT GMBH: Un anticuerpo anti-PD-L1 aislado o su fragmento de union a antigeno que comprende una secuencia de region variable de cadena pesada y de cadena ligera, en donde: […]
Composición para el tratamiento de enfermedades isquémicas o trastornos inflamatorios neurogénicos, que contienen el secretoma de células progenitoras neurales como ingrediente activo, del 22 de Julio de 2020, de S-BIOMEDICS: Una composición para uso en el tratamiento de enfermedad cerebrovascular isquémica, cardiopatía isquémica, infarto de miocardio, enfermedad de Alzheimer, enfermedad […]
Utilización de anticuerpos optimizados en ADCC para tratar a los pacientes con bajo nivel de respuesta, del 22 de Julio de 2020, de LABORATOIRE FRANCAIS DU FRACTIONNEMENT ET DES BIOTECHNOLOGIES: Utilización de una composición de anticuerpo monoclonal quimérico, humanizado o humano de isotipo IgG1 anti- Rhesus del glóbulo rojo humano cuya […]
Composiciones farmacéuticas que contienen una leucocidina E mutada, del 22 de Julio de 2020, de NEW YORK UNIVERSITY: Una composición que comprende: una proteína Leucocidina E (LukE) aislada que comprende la secuencia de aminoácidos de la SEQ ID NO: 4, o un polipéptido […]
Método para producir inmunoconjugados de anticuerpo-SN-38 con un enlazador CL2A, del 22 de Julio de 2020, de IMMUNOMEDICS, INC.: Un método para producir un compuesto, CL2A-SN-38, que presenta la estructura, **(Ver fórmula)** que comprende realizar un esquema de reacción como el que se muestra: **(Ver […]
Anticuerpos del OPGL, del 15 de Julio de 2020, de AMGEN FREMONT INC.: Un anticuerpo, que comprende una cadena pesada y una cadena ligera, donde: a) la cadena pesada comprende: 1) una secuencia de aminoácidos recogida […]