ESTRATEGIAS PARA LA IDENTIFICACIÓN Y DETECCIÓN DE ALTO RENDIMIENTO DE POLIMORFISMOS.
Método para identificar uno o más polimorfismos, comprendiendo dicho método las etapas de:
a) proporcionar una primera muestra de ácidos nucleicos de interés; b) llevar a cabo una reducción de complejidad de la primera muestra de ácidos nucleicos de interés, proporcionando una primera biblioteca de la primera muestra de ácidos nucleicos; c) llevar a cabo consecutiva o simultáneamente las etapas a) y b) con una segunda o posterior muestra de ácidos nucleicos de interés, obteniendo una segunda o posterior biblioteca de la segunda o posterior muestra de ácidos nucleicos de interés; d) secuenciar por lo menos una parte de la primer biblioteca y de la segunda o posterior biblioteca, en la que la secuenciación se lleva a cabo en un soporte sólido, tal como una perla; e) alinear las secuencias obtenidas en la etapa d); f) determinar uno o más polimorfismos entre la primera muestra de ácidos nucleicos y la segunda o posterior muestra de ácidos nucleicos en la alineación de la etapa e); g) utilizar el polimorfismo o polimorfismos determinados en la etapa f) para diseñar sondas de detección; h) proporcionar una muestra de ensayo de ácidos nucleicos de interés; i) llevar a cabo la reducción de complejidad de la etapa b) en la muestra de ensayo de ácidos nucleicos de interés, proporcionando una biblioteca de ensayo de la muestra de ensayo de ácidos nucleicos; j) someter la biblioteca de ensayo a cribado de alto rendimiento para identificar la presencia, ausencia o cantidad de polimorfismos determinados en la etapa f) utilizando las sondas de detección diseñadas en la etapa g), y en la que la reducción de complejidad de la etapa b) se lleva a cabo mediante: - digestión de la muestra de ácidos nucleicos con por lo menos una endonucleasa de restricción para fragmentar en fragmentos de restricción; - ligación de los fragmentos de restricción obtenidos con por lo menos un adaptador oligonucleótido sintético de doble cadena que presenta un extremo compatible con uno o ambos extremos de los fragmentos de restricción, produciendo fragmentos de restricción ligados con adaptadores; - puesta en contacto de dichos fragmentos de restricción ligados con adaptadores con uno o más cebadores oligonucleótidos bajo condiciones de hibridación; y - amplificación de dichos fragmentos de restricción ligados con adaptadores mediante alargamiento de uno o más cebadores oligonucleótidos, - en el que por lo menos uno de entre el cebador o cebadores oligonucleótidos incluye una secuencia de nucleótidos que presenta la misma secuencia de nucleótidos que las partes terminales de las cadenas en los extremos de dichos fragmentos de restricción ligados con adaptadores, incluyendo los nucleótidos implicados en la formación de la secuencia diana para dicha endonucleasa de restricción e incluyendo por lo menos parte de los nucleótidos presentes en los adaptadores, en el que, opcionalmente, por lo menos uno de dichos cebadores incluye en su extremo 3' una secuencia seleccionada que comprende por lo menos un nucleótido inmediatamente contiguo a los nucleótidos implicados en la formación de la secuencia diana de dicha endonucleasa de restricción
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/NL2006/000311.
Solicitante: KEYGENE N.V..
Nacionalidad solicitante: Países Bajos.
Dirección: AGRO BUSINESS PARK 90 6708 PW WAGENINGEN PAISES BAJOS.
Inventor/es: VAN EIJK,MICHAEL,JOSEPHUS,THERESIA, VAN DER POEL,Henricus,Johannes,Adam.
Fecha de Publicación: .
Fecha Solicitud PCT: 23 de Junio de 2006.
Clasificación PCT:
- C12Q1/68 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12Q PROCESOS DE MEDIDA, INVESTIGACION O ANALISIS EN LOS QUE INTERVIENEN ENZIMAS, ÁCIDOS NUCLEICOS O MICROORGANISMOS (ensayos inmunológicos G01N 33/53 ); COMPOSICIONES O PAPELES REACTIVOS PARA ESTE FIN; PROCESOS PARA PREPARAR ESTAS COMPOSICIONES; PROCESOS DE CONTROL SENSIBLES A LAS CONDICIONES DEL MEDIO EN LOS PROCESOS MICROBIOLOGICOS O ENZIMOLOGICOS. › C12Q 1/00 Procesos de medida, investigación o análisis en los que intervienen enzimas, ácidos nucleicos o microorganismos (aparatos de medida, investigación o análisis con medios de medida o detección de las condiciones del medio, p. ej. contadores de colonias, C12M 1/34 ); Composiciones para este fin; Procesos para preparar estas composiciones. › en los que intervienen ácidos nucleicos.
Países PCT: Austria, Bélgica, Suiza, Alemania, Dinamarca, España, Francia, Reino Unido, Grecia, Italia, Liechtensein, Luxemburgo, Países Bajos, Suecia, Mónaco, Portugal, Irlanda, Eslovenia, Finlandia, Rumania, Chipre, Lituania, Letonia.
PDF original: ES-2357549_T3.pdf
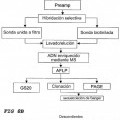



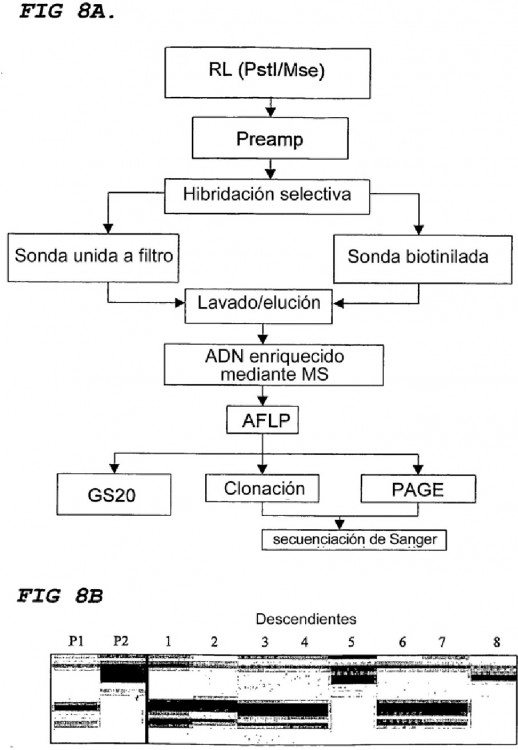
Fragmento de la descripción:
Campo técnico
La presente invención se refiere a los campos de la biología molecular y de la genética. La invención se refiere a la rápida identificación de múltiples polimorfismos en una muestra de ácidos nucleicos. Los polimorfismos identificados pueden utilizarse para el desarrollo de sistemas de cribado de alto rendimiento para polimorfismos en muestras de ensayo.
Antecedentes de la invención
La exploración del ADN genómico ha sido el deseo de la comunidad científica, en particular de la comunidad médica, desde hace mucho tiempo. El ADN genómico es la clave para la identificación, diagnóstico y tratamiento de enfermedades tales como el cáncer y la enfermedad de Alzheimer. Además de la identificación y tratamiento de enfermedades, la exploración del ADN genómico podría proporcionar ventajas significativas en esfuerzos de cría vegetal y animal, proporcionando respuestas a problemas de alimentación y nutrición en todo el mundo. Es conocido que muchas enfermedades se asocian a componentes genéticos específicos, en particular a polimorfismos en genes específicos. La identificación de polimorfismos en muestras grandes, tales como genomas, en la actualidad es una tarea laboriosa y que requiere mucho tiempo. Sin embargo, esta identificación resulta de gran valor para áreas tales como la investigación biomédica, el desarrollo de productos farmacéuticos, el tipado de tejidos, el genotipado y los estudios poblacionales.
La patente WO 2004/022758 describe un método para la fragmentación del genoma que utiliza una pluralidad de enzimas de restricción para producir una fragmentación representativa de un ácido nucleico de muestra ligado en un vector con el fin de proporcionar una biblioteca que contenga los fragmentos.
Nicod et al., Nucleic acids research 31(5):19, 1 de marzo de 2003, describe un método para identificar SNPs basándose en la AFLP utilizando geles radioactivos, la extracción uno a uno de los fragmentos de la AFLP y la evitación de la selección de fragmentos que se encuentran presentes en todos los individuos investigados.
Descripción resumida de la invención
La presente invención proporciona un método para identificar eficientemente y para detectar fiablemente polimorfismos en una muestra de ácidos nucleicos (por ejemplo ADN o ARN) compleja, por ejemplo muy grande, de un modo rápido y económico utilizando una combinación de métodos de alto rendimiento.
Dicha integración de métodos de alto rendimiento conjuntamente proporcionan una plataforma que resulta particularmente adecuada para la identificación y detección rápidas y fiables de polimorfismos en muestras de ácidos nucleicos altamente complejas, en las que la identificación y mapeado convencionales de polimorfismos resultaría laboriosa y que requeriría mucho tiempo.
Una de la cosas que han encontrado los presentes inventores es una solución para identificar polimorfismos, preferentemente polimorfismos de un solo nucleótido, aunque de manera similar para (micro)satélites y/o indels, en particular en genomas grandes. El método es único en su aplicabilidad a genomas tanto grandes como pequeños, aunque proporciona ventajas particulares en genomas grandes, en particular en especies poliploides.
Para identificar los SNPs (y posteriormente detectar los SNPs identificados) se dispone de varias posibilidades en la técnica. En una primera opción, puede secuenciarse el genoma completo, y ello puede llevarse a cabo en varios individuos. Es un ejercicio en gran parte teórico, al ser engorroso y caro y, a pesar del rápido desarrollo de la tecnología, simplemente no resulta factible su aplicación a cada organismo, especialmente a los que presentan genomas más grandes. La segunda opción es utilizar la información de secuencia disponible (fragmentada), tal como las bibliotecas EST. Esto permite la generación de cebadores de PCR, la resecuenciación y la comparación entre individuos. Nuevamente lo anterior requiere información inicial de secuencia que no se encuentra disponible o que se encuentra disponible sólo en una cantidad limitada. Además, deben desarrollarse ensayos de PCR separados para cada región, lo que supone una adición enorme de costes y tiempo de desarrollo.
La tercera opción es limitarse a parte del genoma de cada individuo. La dificultad reside en que la parte proporcionada del genoma debe ser igual en diferentes individuos con el fin de proporcionar un resultado comparable para la identificación con éxito de SNPs. Los presentes inventores ahora han resuelto este dilema mediante la integración de métodos altamente reproducibles para seleccionar parte del genoma mediante secuenciación de alto rendimiento para la identificación de los polimorfismos integrada con la preparación de muestras y plataformas de identificación de alto rendimiento. La presente invención acelera el procedimiento de identificación de polimorfismos y utiliza los mismos elementos en el procedimiento posterior para explotar los polimorfismos descubiertos, permitiendo un genotipado de alto rendimiento efectivo y fiable.
Entre las aplicaciones adicionalmente contempladas por la presente invención se incluyen las bibliotecas de microsatélites enriquecidas mediante cribado, la realización de AFLP-ADNc de perfilado de transcritos (northern digital), la secuenciación de genomas complejos, la secuenciación de bibliotecas de EST (en ADNc completo o en AFLP-ADNc), la exploración de microARN (secuenciación de bibliotecas de inserciones de pequeño tamaño), la secuenciación de cromosoma artificial bacteriano (BAC) (contig), AFLP/AFLP-ADNc en un enfoque de análisis de segregantes agrupados, la detección rutinaria de fragmentos de la AFLP, por ejemplo para retrocruzamientos asistidos por un marcador (MABC), etc.
Definiciones
En la descripción y ejemplos posteriormente se utiliza una serie de expresiones. Con el fin de proporcionar una comprensión clara y consistente de la memoria y reivindicaciones, incluyendo el alcance que debe proporcionarse a dichas expresiones, se proporcionan las definiciones siguientes. A menos que se defina de otra manera en la presente memoria, todas las expresiones técnicas y científicas utilizadas presentan los mismos significados comúnmente entendidos por el experto ordinario en la materia a la que pertenece la presente invención.
Polimorfismo: los polimorfismos se refieren a la presencia de dos o más variantes de una secuencia de nucleótidos en una población. Un polimorfismo puede comprender uno o más cambios de bases, una inserción, una repetición, o una deleción. Un polimorfismo incluye, por ejemplo una repetición de secuencia simple (SSR) y un polimorfismo de un único nucleótidos (SNP), que es una variación que se produce en el caso de que se altere un único nucleótido: adenina (A), timina (T), citosina (C) o guanina (G). Debe producirse generalmente una variación en por lo menos 1% de la población para que se considere un SNP. Los SNPs constituyen 90% de todas las variaciones genéticas humanas y se producen cada 100 a 300 bases a lo largo del genoma humano. Dos de cada tres SNPs sustituyen la citosina (C) por la timina (T). Las variaciones en las secuencias de ADN de, por ejemplo, seres humanos o plantas, pueden afectar a cómo se enfrentan a enfermedades, bacterias, virus, compuestos químicos, fármacos, etc.
Ácido nucleico: un ácido nucleico según la presente invención puede incluir cualquier polímero u oligómero de bases pirimidina o purina, preferentemente citosina, timina, y uracilo,y adenina y guanina, respectivamente (ver Albert L. Lehninger, Principles of Biochemistry, páginas 793 a 800, Worth Publ, 1982). La presente invención contempla cualquier componente desoxirribonucleótido, ribonucleótido o péptido-ácido nucleico, y cualesquiera variantes químicas de los mismos, tales como formas metiladas, hidroximetiladas o glucosiladas de dichas bases, y similares. Los polímeros u oligómeros pueden ser de composición heterogénea u homogénea, y pueden aislarse a partir de fuentes naturales o producirse artificial o sintéticamente. Además, los ácidos nucleicos pueden ser de ADN o ARN, o una mezcla de los mismos, y pueden existir permanente o transitoriamente en forma de una cadena o de doble cadena, incluyendo estados de homodúplex, heterodúplex e híbridos.
Reducción de complejidad: la expresión "reducción de la complejidad" se utiliza para referirse a un método en el que la complejidad de una muestra de ácidos nucleicos, tal como ADN genómico, se reduce mediante la generación... [Seguir leyendo]
Reivindicaciones:
1. Método para identificar uno o más polimorfismos, comprendiendo dicho método las etapas de:
a) proporcionar una primera muestra de ácidos nucleicos de interés;
b) llevar a cabo una reducción de complejidad de la primera muestra de ácidos nucleicos de interés, proporcionando una primera biblioteca de la primera muestra de ácidos nucleicos;
c) llevar a cabo consecutiva o simultáneamente las etapas a) y b) con una segunda o posterior muestra de ácidos nucleicos de interés, obteniendo una segunda o posterior biblioteca de la segunda o posterior muestra de ácidos nucleicos de interés;
d) secuenciar por lo menos una parte de la primer biblioteca y de la segunda o posterior biblioteca, en la que la secuenciación se lleva a cabo en un soporte sólido, tal como una perla;
e) alinear las secuencias obtenidas en la etapa d);
f) determinar uno o más polimorfismos entre la primera muestra de ácidos nucleicos y la segunda o posterior muestra de ácidos nucleicos en la alineación de la etapa e);
g) utilizar el polimorfismo o polimorfismos determinados en la etapa f) para diseñar sondas de detección;
h) proporcionar una muestra de ensayo de ácidos nucleicos de interés;
i) llevar a cabo la reducción de complejidad de la etapa b) en la muestra de ensayo de ácidos nucleicos de interés, proporcionando una biblioteca de ensayo de la muestra de ensayo de ácidos nucleicos;
j) someter la biblioteca de ensayo a cribado de alto rendimiento para identificar la presencia, ausencia o cantidad de polimorfismos determinados en la etapa f) utilizando las sondas de detección diseñadas en la etapa g), y en la que la reducción de complejidad de la etapa b) se lleva a cabo mediante:
- digestión de la muestra de ácidos nucleicos con por lo menos una endonucleasa de restricción para fragmentar en fragmentos de restricción;
- ligación de los fragmentos de restricción obtenidos con por lo menos un adaptador oligonucleótido sintético de doble cadena que presenta un extremo compatible con uno o ambos extremos de los fragmentos de restricción, produciendo fragmentos de restricción ligados con adaptadores;
- puesta en contacto de dichos fragmentos de restricción ligados con adaptadores con uno o más cebadores oligonucleótidos bajo condiciones de hibridación; y
- amplificación de dichos fragmentos de restricción ligados con adaptadores mediante alargamiento de uno o más cebadores oligonucleótidos,
- en el que por lo menos uno de entre el cebador o cebadores oligonucleótidos incluye una secuencia de nucleótidos que presenta la misma secuencia de nucleótidos que las partes terminales de las cadenas en los extremos de dichos fragmentos de restricción ligados con adaptadores, incluyendo los nucleótidos implicados en la formación de la secuencia diana para dicha endonucleasa de restricción e incluyendo por lo menos parte de los nucleótidos presentes en los adaptadores, en el que, opcionalmente, por lo menos uno de dichos cebadores incluye en su extremo 3' una secuencia seleccionada que comprende por lo menos un nucleótido inmediatamente contiguo a los nucleótidos implicados en la formación de la secuencia diana de dicha endonucleasa de restricción.
2. Método según la reivindicación 1, en el que el adaptador y/o el cebador comprende una etiqueta.
3. Método según la reivindicación 2, en el que la etiqueta es una secuencia identificadora.
4. Método según la reivindicación 1, en el que por lo menos uno de los cebadores se encuentra fosforilado.
5. Método según cualquiera de las reivindicaciones anteriores, en el que la secuenciación se basa en la secuenciación mediante terminación de cadena dideoxi.
6. Método según la reivindicación 1, en el que la secuenciación comprende las etapas de:
- unir los fragmentos ligados con adaptadores a perlas, uniendo cada perla con un único fragmento ligado con adaptador;
- emulsionar las perlas en microrreactores de agua en aceite, comprendiendo cada microrreactor de agua en aceite una única perla;
- cargar las perlas en pocillos, comprendiendo cada pocillo una única perla; y
- generar una señal pirofosfato.
7. Método según la reivindicación 6, en el que, antes de la etapa de unión, se ligan adaptadores de secuenciación con los fragmentos dentro de la primera biblioteca etiquetada y de la segunda biblioteca etiquetada o la biblioteca combinada.
8. Método según la reivindicación 7, en el que los adaptadores de secuenciación portan un extremo protuberante 3'-T.
9. Método según cualquiera de las reivindicaciones anteriores, en el que el cribado de alto rendimiento se lleva a cabo mediante inmovilización de las sondas diseñadas en la etapa h) sobre una matriz, seguido de la puesta en contacto de la matriz que comprende las sondas con una biblioteca de ensayo bajo condiciones de hibridación.
10. Método para identificar uno o más polimorfismos, comprendiendo dicho método las etapas de:
a) proporcionar una pluralidad de muestras de ácidos nucleicos de interés,
b) llevar a cabo una reducción de complejidad de cada una de las muestras para proporcionar una pluralidad de bibliotecas de las muestras de ácidos nucleicos, en la que la reducción de complejidad se lleva a cabo mediante:
- digestión de cada muestra de ácidos nucleicos con por lo menos una endonucleasa de restricción para fragmentarla en fragmentos de restricción;
- ligación de los fragmentos de restricción obtenidos con por lo menos un adaptador oligonucleótido sintético de doble cadena que presenta un extremo compatible con uno o con ambos extremos de los fragmentos de restricción, produciendo fragmentos de restricción ligados con adaptadores;
- puesta en contacto de dichos fragmentos de restricción ligados con adaptadores, con uno o más cebadores oligonucleótidos fosforilados bajo condiciones de hibridación; y
- amplificación de dichos fragmentos de restricción ligados con adaptadores mediante alargamiento de uno o más de los cebadores oligonucleótidos, en la que por lo menos uno de entre el cebador o cebadores oligonucleótidos incluye una secuencia de nucleótidos que presenta la misma secuencia de nucleótidos que las partes terminales de las cadenas en los extremos de dichos fragmentos de restricción ligados con adaptadores, incluyendo los nucleótidos implicados en la formación de la secuencia diana para dicha endonucleasa de restricción, e incluyendo por lo menos parte de los nucleótidos presentes en los adaptadores, en el que, opcionalmente, por lo menos uno de dichos cebadores incluye en su extremo 3' una secuencia seleccionada que comprende por lo menos un nucleótido situado inmediatamente contiguo a los nucleótidos implicados en la formación de la secuencia diana para dicha endonucleasa de restricción y en el que el adaptador y/o el cebador contiene una etiqueta;
c) combinación de dichas bibliotecas para formar una biblioteca combinada;
d) ligar los adaptadores de secuenciación capaces de unirse a perlas, con los fragmentos con caperuza de adaptador amplificados en la biblioteca combinada, utilizando un adaptador de secuenciación que porta un extremo protuberante 3'-T, y someter los fragmentos unidos a perla a polimerización en emulsión;
e) secuenciar por lo menos una parte de la biblioteca combinada;
f) alinear las secuencias de cada muestra obtenidas en la etapa e);
g) determinar uno o más polimorfismos entre la pluralidad de muestras de ácidos nucleicos en la alineación de la etapa f);
h) utilizar uno o más polimorfismos determinados en la etapa g) para diseñar sondas de detección;
i) proporcionar una muestra de ensayo de ácidos nucleicos de interés;
j) llevar a cabo la reducción de complejidad de la etapa b) en la muestra de ensayo de ácidos nucleicos de interés, proporcionando una biblioteca de ensayo de la muestra de ensayo de ácidos nucleicos;
k) someter la biblioteca de ensayo a cribado de alto rendimiento para identificar la presencia, ausencia o cantidad de polimorfismos determinada en la etapa g) utilizando las sondas de detección diseñadas en la etapa h). 5 11. Utilización de los métodos según las reivindicaciones 1 a 6 para el cribado de bibliotecas de microsatélites enriquecidas, la realización de AFLP-ADNc de perfilado de transcritos (northern digital), la secuenciación de genomas complejos, la secuenciación de bibliotecas de etiquetas de secuencia expresadas (en ADNc completo o en ADNc-AFLP), la exploración de microARN (secuenciación de bibliotecas de inserciones de pequeño tamaño), la secuenciación de cromosomas artificiales 10 bacterianos (BACs), el enfoque de análisis de segregantes agrupados en combinación con AFLP/AFLP-ADNc y la detección rutinaria de fragmentos de la AFLP (retrocruzamientos asistidos por un marcador).
Patentes similares o relacionadas:
Método para analizar ácido nucleico molde, método para analizar sustancia objetivo, kit de análisis para ácido nucleico molde o sustancia objetivo y analizador para ácido nucleico molde o sustancia objetivo, del 29 de Julio de 2020, de Kabushiki Kaisha DNAFORM: Un método para analizar un ácido nucleico molde, que comprende las etapas de: fraccionar una muestra que comprende un ácido nucleico molde […]
MÉTODOS PARA EL DIAGNÓSTICO DE ENFERMOS ATÓPICOS SENSIBLES A COMPONENTES ALERGÉNICOS DEL POLEN DE OLEA EUROPAEA (OLIVO), del 23 de Julio de 2020, de SERVICIO ANDALUZ DE SALUD: Biomarcadores y método para el diagnostico, estratificación, seguimiento y pronostico de la evolución de la enfermedad alérgica a polen del olivo, kit […]
Detección de interacciones proteína a proteína, del 15 de Julio de 2020, de THE GOVERNING COUNCIL OF THE UNIVERSITY OF TORONTO: Un método para medir cuantitativamente la fuerza y la afinidad de una interacción entre una primera proteína de membrana o parte de la misma y una […]
Secuenciación dirigida y filtrado de UID, del 15 de Julio de 2020, de F. HOFFMANN-LA ROCHE AG: Un procedimiento para generar una biblioteca de polinucleótidos que comprende: (a) generar una primera secuencia del complemento (CS) de un polinucleótido diana a partir […]
Métodos para la recopilación, estabilización y conservación de muestras, del 8 de Julio de 2020, de Drawbridge Health, Inc: Un método para estabilizar uno o más componentes biológicos de una muestra biológica de un sujeto, comprendiendo el método obtener un […]
Evento de maíz DP-004114-3 y métodos para la detección del mismo, del 1 de Julio de 2020, de PIONEER HI-BRED INTERNATIONAL, INC.: Un amplicón que consiste en la secuencia de ácido nucleico de la SEQ ID NO: 32 o el complemento de longitud completa del mismo.
Composiciones para modular la expresión de SOD-1, del 24 de Junio de 2020, de Biogen MA Inc: Un compuesto antisentido según la siguiente fórmula: mCes Aeo Ges Geo Aes Tds Ads mCds Ads Tds Tds Tds mCds Tds Ads mCeo Aes Geo mCes Te (secuencia […]
Aislamiento de ácidos nucleicos, del 24 de Junio de 2020, de REVOLUGEN LIMITED: Un método de aislamiento de ácidos nucleicos que comprenden ADN de material biológico, comprendiendo el método las etapas que consisten en: (i) efectuar un lisado […]