48 patentes, modelos y diseños de HANMI PHARM. CO., LTD.
Formulación granular compleja con estabilidad mejorada, que comprende levocetirizina y montelukast.
Sección de la CIP Necesidades corrientes de la vida
(13/05/2020). Inventor/es: WOO, JONG SOO, PARK,JAE HYUN, KIM,YONG IL, IM,HO TAEK, KWON,TEAK KWAN. Clasificación: A61K31/495, A61K9/16, A61K31/4704, A61K47/40.
Una formulación granular compleja que comprende:
(a) una primera parte granular que comprende levocetirizina o una sal farmacéuticamente aceptable de la misma, ciclodextrina o un derivado de la misma y un agente alcalinizante; y
(b) una segunda parte granular que comprende montelukast o una sal farmacéuticamente aceptable del mismo, ciclodextrina o un derivado de la misma y un agente alcalinizante,
en donde el agente alcalinizante de la primera y segunda partes granulares se selecciona del grupo que consiste en meglumina, bicarbonato sódico, carbonato sódico monohidrato, agua amoniacal, citrato sódico, carbonato sódico seco y una mezcla de los mismos, y
en donde la primera y segunda partes granulares contienen la ciclodextrina o su derivado en una cantidad del 3 al 7 % en peso basado en el peso total de cada parte granular.
PDF original: ES-2803512_T3.pdf
Un conjugado de fc de inmunoglobulina que mantiene la afinidad de unión del fragmento fc de inmunoglobulina a fcrn.
(18/03/2020) Una composición, que comprende un conjugado de fragmento Fc de inmunoglobulina-polipéptido fisiológicamente activo que mantiene la afinidad de unión intrínseca del fragmento Fc de inmunoglobulina por FcRn,
el conjugado que comprende solo una molécula de un polipéptido fisiológicamente activo unido mediante un enlazador no peptidilo a un fragmento Fc de inmunoglobulina que comprende una región de unión a FcRn,
en donde una relación de unión del conjugado está dentro del intervalo de ± 6 % de la relación de unión del fragmento Fc de inmunoglobulina determinada en las mismas condiciones que las usadas para el conjugado, en donde la relación de unión del conjugado y la relación de unión del fragmento Fc de inmunoglobulina se determinan mediante el uso de la siguiente ecuación:
Relación de unión (%) = (cantidad unida…
Una composición para el tratamiento de la diabetes que comprende un análogo de oxintomodulina.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(11/03/2020). Inventor/es: KWON, SE CHANG, JUNG, SUNG YOUB, KIM,DAE-JIN, KIM,JIN SUN, LEE,SANG HYUN. Clasificación: A61K38/22, C07K14/575, C07K16/30, C07K14/605, A61K47/64.
Una composición para su uso en la prevención o el tratamiento de la diabetes, la composición comprende un conjugado de análogo de oxintomodulina como un ingrediente activo en donde el conjugado de análogo de oxintomodulina comprende
un análogo de oxintomodulina que comprende la secuencia de aminoácidos de las SEQ ID NO: 24, 25 o 26;
una región Fc de inmunoglobulina; y
un polímero no peptidílico, en donde el polímero no peptidílico une el análogo de oxintomodulina a la región Fc de inmunoglobulina a través de un enlace covalente.
PDF original: ES-2802099_T3.pdf
Formulación compuesta, que comprende un comprimido esferoidal de múltiples unidades (MUST) encapsulado en una cápsula dura y un método para preparar el mismo.
Sección de la CIP Necesidades corrientes de la vida
(19/02/2020). Inventor/es: WOO, JONG SOO, PARK,JAE HYUN, KIM,KYEONG SOO, KIM,YONG IL, KWON,TAEK KWAN, KIM,DONG HO. Clasificación: A61K31/505, A61K31/495, A61K31/47, A61K31/137, A61K31/4178, A61K9/48, A61K31/496, A61K31/616, A61K31/4422, A61K31/4365.
Una formulación compuesta de cápsula dura, que comprende dos o más principios farmacéuticamente activos, en donde cada principio farmacéuticamente activo está contenido en un comprimido esferoidal de múltiples unidades (MUST), que tiene un diámetro en un intervalo de 1 a 4 mm, y de 4 a 40 MUST por cada principio farmacéuticamente activo están encapsulados en la cápsula dura; y en donde cada MUST tiene una relación entre diámetro y espesor en un intervalo de 1:0,7 a 1:1,3 y una relación entre espesor y altura de cilindro en un intervalo de 1:0,3 a 1:0,9.
PDF original: ES-2778864_T3.pdf
Preparación de material compuesto que comprende una capa de recubrimiento de película, que contiene un inhibidor de la 5-alfa-reductasa, y método para la producción de la preparación de material compuesto.
(19/02/2020) Una preparación de material compuesto, que comprende:
un núcleo, que contiene un primer principio activo; y
una capa de recubrimiento de película, que contiene un inhibidor de la 5-α-reductasa,
en donde se forma la capa de recubrimiento de película, mediante el recubrimiento con una solución de recubrimiento de película, que comprende el inhibidor de la 5-α-reductasa y un material de recubrimiento de película en un disolvente mixto de agua y del 30 % en peso al 80 % en peso de un disolvente orgánico con respecto al peso total del disolvente mixto, en donde el material de recubrimiento de película es una combinación de un copolímero de injerto de alcohol de polivinilo-polietilen glicol y alcohol de polivinilo,
en donde la relación en peso del copolímero de injerto de alcohol de polivinilo-polietilen glicol respecto…
Preparación de composite que comprende una capa de recubrimiento pelicular que contiene principio activo.
Sección de la CIP Necesidades corrientes de la vida
(08/01/2020). Inventor/es: WOO, JONG SOO, CHO,JUNG HYUN, PARK,JAE HYUN, KIM,KYEONG SOO, KIM,YONG IL, KIM,JIN CHEUL, CHOI,YOUNG KEUN, KIM,HYUNG SEO. Clasificación: A61K9/20, A61K45/06, A61K47/32, A61K9/48, A61K47/30.
Una preparación de composite que comprende:
un núcleo que contiene un primer principio activo; y
una capa de recubrimiento pelicular que contiene un segundo principio activo,
en donde la capa de recubrimiento pelicular comprende un copolímero de injerto de poli(alcohol vinílico)- polietilenglicol y poli(alcohol vinílico),
en donde una relación en peso entre el copolímero de injerto de poli(alcohol vinílico)-polietilenglicol y el poli(alcohol vinílico) es de 7:3 a 4:6, y
en donde el segundo principio activo comprende un inhibidor de la 5-α-reductasa seleccionado del grupo que consiste en finasterida, dutasterida y cualquier combinación de los mismos.
PDF original: ES-2772137_T3.pdf
Análogo de la insulina y uso del mismo.
Secciones de la CIP Química y metalurgia Necesidades corrientes de la vida
(13/11/2019). Inventor/es: KWON, SE CHANG, JUNG, SUNG YOUB, CHOI,IN-YOUNG, KIM,DAE-JIN, KIM,JIN YOUNG, HONG,SUNG HEE, HUH,YONG HO, JANG,MYUNG HYUN, KIM,SEUNG SU, HWANG,SANG YOUN, KIM,HYUN UK. Clasificación: C07K14/62, A61K47/68.
Un análogo de la insulina que tiene un título de insulina reducido en comparación con la forma nativa, en el que el análogo de la insulina tiene una cadena A de SEQ ID NO: 37 y cadena B de SEQ ID NO: 38 excepto por una sustitución del 14to aminoácido (tirosina) de la cadena A con ácido glutámico o asparagina.
PDF original: ES-2770776_T3.pdf
Nuevo método para la preparación de un compuesto de tienopirimidina e intermedios usados en el mismo.
Sección de la CIP Química y metalurgia
(16/10/2019). Inventor/es: SUH, KWEE, HYUN, HA, TAE, HEE, JUNG,JAE HYUK, KIM,HO SEOK, BAEK,JONG OUK. Clasificación: C07D495/04, C07D417/12, C07D403/12.
Un método para preparar un compuesto de Fórmula 1, que comprende las etapas de:
a. permitir que un compuesto de Fórmula 3 o una sal del mismo reaccionen con un agente de cloración para obtener un compuesto de Fórmula 2 o una sal del mismo; y
b. permitir que el compuesto de Fórmula 2 o la sal del mismo reaccionen con un compuesto de Fórmula 8 o una sal del mismo y una base:**Fórmula**.
PDF original: ES-2759795_T3.pdf
Derivados de tieno[3,2-d]pirimidina que tienen actividad inhibitoria para las proteínas quinasas.
(25/09/2019) Un derivado de tieno[3,2-d]pirimidina de fórmula (I):**Fórmula**
en la que,
A es arilo C6-10 o heteroarilo de 5 a 10 miembros;
W es O, S, S(O), S(O)2, NH, -NHNH- o heterocicloalquilo de 3 a 6 miembros;
X y Y son cada uno independientemente CH o N;
Z es hidrógeno, alquilo C1-3 o NR3R4, en donde dicho R3 y R4 son cada uno independientemente hidrógeno, alquilo C1- 6 o -(CH2)q-B, B que representan NR5R6, alcoxi C1-6, cicloalquilo C3-6 o heterocicloalquilo de 3 a 6 miembros;
R1 es hidrógeno, halógeno, alquilo C1-3 o alcoxi C1-3, en donde dicho alquilo o alcoxi no está sustituido o está sustituido con uno o más átomos…
Una formulación líquida del conjugado de péptido insulinotrópico de acción prolongada.
Sección de la CIP Necesidades corrientes de la vida
(11/09/2019). Inventor/es: KWON, SE CHANG, BAE, SUNG MIN, KIM,DAE-JIN, HONG,SUNG HEE, KIM,HYUN UK, LIM,HYUNG KYU. Clasificación: A61K38/28, A61K9/08, A61K38/26.
Una formulación líquida del conjugado de péptido insulinotrópico de acción prolongada, que comprende una cantidad con eficacia farmacéutica del conjugado de péptido insulinotrópico de acción prolongada en la que un péptido insulinotrópico, que es un péptido activo fisiológicamente, se une a una región Fc de inmunoglobulina; y un estabilizador libre de albúmina, en la que el estabilizador comprende un tampón, un alcohol de azúcar, y un tensioactivo no iónico; y
en la que el péptido insulinotrópico es exendina-3, exendina-4, o imidazo-acetil exendina-4; y
en la que el tampón es un tampón citrato, el alcohol de azúcar es manitol o sacarosa, y el tensioactivo no iónico es polisorbato.
PDF original: ES-2759987_T3.pdf
Dispersión sólida amorfa que comprende taxano, comprimido que comprende la misma y método de preparación de los mismos.
Sección de la CIP Necesidades corrientes de la vida
(14/08/2019). Inventor/es: WOO, JONG SOO, PARK,JAE HYUN, KIM,YONG IL, IM,HO TAEK, SRINIVASAN,SHANMUGAM, YOON,YOUNG SU. Clasificación: A61K31/335, A61K9/20, A61K9/16, A61K9/14, A61K31/337, A61K9/30.
Una dispersión sólida amorfa que comprende un taxano o una sal farmacéuticamente aceptable del mismo, polivinilpirrolidona, polisorbato y laurilsulfato de sodio.
PDF original: ES-2748686_T3.pdf
Formulación líquida de conjugado de proteínas que comprende la oxintomodulina y un fragmento de inmunoglobulina.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(10/07/2019). Inventor/es: KWON, SE CHANG, BAE, SUNG MIN, JANG,MYUNG HYUN, KIM,HYUN UK, LIM,HYUNG KYU, KIM,SANG YUN. Clasificación: A61K47/12, A61K9/00, A61K47/10, A61K47/26, A61K9/08, A61K38/22, C07K14/605, A61K38/26, A61K47/68.
Una formulación líquida de un conjugado derivado de oxintomodulina de larga duración, que comprende una cantidad farmacológicamente activa de un conjugado derivado de oxintomodulina de larga duración en la que el conjugado derivado de oxintomodulina comprende un derivado de oxintomodulina, que es un péptido fisiológicamente activo, que comprende la secuencia de aminoácidos de una cualquiera de las SEQ ID NO: 24, 25 o 26; una región Fc de inmunoglobulina; y
un polímero no peptidílico, en la que el polímero no peptidílico une covalentemente el derivado de oxintomodulina y la región Fc de inmunoglobulina,
y un estabilizador sin albúmina, en la que el estabilizador contiene un tampón que tiene un pH que varía de 4,8 a 6,0, uno o más alcoholes de azúcar seleccionados de manitol y sorbitol y polisorbato 20.
PDF original: ES-2748158_T3.pdf
Una formulación líquida de insulina y péptido insulinotrópico de acción prolongada.
Sección de la CIP Necesidades corrientes de la vida
(05/06/2019). Inventor/es: KWON, SE CHANG, BAE, SUNG MIN, LEE,JONG-SOO, LEE MI KYOUNG, KIM,HYUN UK, LIM,HYUNG KYU. Clasificación: A61K38/18, A61K38/28, A61K9/08.
Una formulación líquida de una combinación de conjugado de insulina de acción prolongada y conjugado de péptido insulinotrópico de acción prolongada, que comprende:
un conjugado de insulina de acción prolongada en el que la insulina está enlazada a la región Fc de inmunoglobulina,
un conjugado de péptido insulinotrópico de acción prolongada en el que el péptido insulinotrópico está enlazado a la región Fc de inmunoglobulina, y
un estabilizador libre de albúmina, en el que el estabilizador comprende un tampón de intervalo de pH de 5,0 a 7,0, un alcohol de azúcar, un tensioactivo no iónico y un agente isotónico;
en la que el péptido insulinotrópico es un péptido similar al glucagón 1 (GLP-1), péptido similar al glucagón 2 (GLP-2), exendina-3, o exendina-4, o un derivado estructural de los mismos.
PDF original: ES-2743918_T3.pdf
Procedimiento de preparación mejorado para producción de alto rendimiento de un conjugado de polipéptido fisiológicamente activo.
(05/06/2019) Un procedimiento para preparar un conjugado de polipéptido fisiológicamente activo - polímero no peptidilo - región constante de inmunoglobulina, que comprende las etapas de:
reacción de un polímero no peptidilo con uno de un polipéptido fisiológicamente activo o una región constante de inmunoglobulina para preparar un complejo de polipéptido fisiológicamente activo - polímero no peptidilo o un complejo de la región constante de inmunoglobulina - polímero no peptidilo; y
reacción del complejo del polipéptido fisiológicamente activo - polímero no peptidilo o el complejo de la región constante de inmunoglobulina - polímero…
Formulación liquida de conjugado de insulina de acción prolongada.
Sección de la CIP Necesidades corrientes de la vida
(03/06/2019). Inventor/es: KWON, SE CHANG, BAE, SUNG MIN, KIM,MIN-YOUNG, HONG,SUNG HEE, KIM,HYUN UK, LIM,HYUNG KYU. Clasificación: A61K38/28, A61K47/12, A61K47/10, A61K47/26, A61K9/08, A61K38/26, A61K47/68.
Una formulación liquida de conjugado de insulina de acción prolongada que comprende una cantidad farmacéuticamente eficaz de un conjuntado de insulina de acción prolongada, en el que una insulina, que es un péptido fisiológicamente activo, se une a una región Fc de inmunoglobulina; y un estabilizante desprovisto de albúmina, en el que el estabilizante comprende un tampón, un alcohol de azúcar, un tensioactivo no iónico y un agente isotónico, en la que:
el tampón es tampón acetato que tiene un pH comprendido entre 5,6 y 7,0;
el alcohol de azúcar es manitol o sacarosa;
el tensioactivo no iónico es polisorbato que tiene una concentración de 0,001 a 0,02 % (p/v); y
el agente isotónico es cloruro sódico.
PDF original: ES-2715326_T3.pdf
Conjugado de insulina específico de sitio.
Secciones de la CIP Química y metalurgia Necesidades corrientes de la vida
(15/05/2019). Inventor/es: KWON, SE CHANG, JUNG, SUNG YOUB, KIM,DAE-JIN, JANG,MYUNG HYUN, HWANG,SANG YOUN, KIM,HYUN UK. Clasificación: C07K14/62, A61K38/28, A61P3/10, C07K17/08, A61K47/68.
Un conjugado de insulina, en el que la insulina y una región Fc de inmunoglobulina están ligadas entre sí a través de un ligador polimérico no peptídico seleccionado del grupo que consiste en polietilenglicol, polipropilenglicol, un copolímero de etilenglicol-propilenglicol, poliol polioxietilado, alcohol polivinílico, polisacárido, dextrano, polivinil etil éter, un polímero biodegradable, un polímero lipídico, quitina, ácido hialurónico y una combinación de los mismos, y un extremo del polímero no peptídico está ligado a cualquiera de los residuos de aminoácidos en las posiciones 20 a 29 de una cadena beta de insulina y el otro extremo de la misma está ligado a la región Fc de inmunoglobulina; en el que la insulina es una insulina nativa o tiene al menos un 80% de homología con la insulina nativa.
PDF original: ES-2738676_T3.pdf
Dispersión sólida con solubilidad mejorada que comprende derivado de tetrazol como principio activo.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(24/04/2019). Inventor/es: WOO, JONG SOO, CHOI,JUN YOUNG, PARK,JAE HYUN, KIM,YONG IL, CHOI,YOUNG KEUN. Clasificación: A61P35/00, C07D405/14, C07D257/04, A61K31/4725.
Una dispersión sólida amorfa que comprende un derivado de tetrazol de fórmula (I) o una sal farmacéuticamente aceptable del mismo como principio activo y un polímero soluble en agua:**Fórmula**
en la que,
R1 es quinolina, isoquinolina, quinoxalina, piridina, pirazina, naftaleno, fenilo, tiofeno, furano, 4-oxo-4H-cromeno o cinolina, que está sin sustituir o sustituido con alquilo C1-C5, hidroxilo, alcoxi C1-5, halógeno, trifluorometilo, nitro o amino;
R2 a R5 y R8 a R11 son cada uno independientemente H, hidroxilo, halógeno, nitro, alquilo C1-C5 o alcoxi C1-5; R6 y R7 son cada uno independientemente H, hidroxilo, halógeno, nitro, alquileno C1-5 o alcoxi C1-5; y R6 y R7 pueden estar conectados para formar un anillo de 4 a 8 miembros;
m y n son cada uno independientemente números enteros que varían de 0 a 4; y
X es CH2, O o S.
PDF original: ES-2731806_T3.pdf
Formulación de cápsula que comprende Montelukast y Levocetirizina.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(24/04/2019). Inventor/es: WOO, JONG SOO, PARK,JAE HYUN, KIM,KYEONG SOO, KIM,YONG IL, KWON,TAEK KWAN, KIM,DONG HO. Clasificación: A61K31/47, A61K9/48, A61K31/4704, C07D215/18.
Una formulación de cápsula para prevenir o tratar rinitis alérgica y asma, que comprende dos capas por separado de:
una capa de Montelukast que comprende montelukast o una sal farmacéuticamente aceptable del mismo; y
una capa de Levocetirizina que comprende levocetirizina o una sal farmacéuticamente aceptable de la misma, en donde dicha capa de Montelukast y dicha capa de Levocetirizina contiene agua en una cantidad del 5 % o menos.
PDF original: ES-2727861_T3.pdf
Derivados de tienopirimidina que tienen actividad inhibidora para la proteína cinasa.
(17/04/2019) Un compuesto que se selecciona entre el grupo que consiste en un derivado de heteroarilo bicíclico de fórmula (I), una sal del mismo, un hidrato del mismo y un solvato del mismo farmacéuticamente aceptables:
en donde,
W es CH;
X es S;
Y es -(CH2)2R3, -CHCR2R3, -CCR3, -C(O)OR3, -C(O)OH o
C(O)NR2R3;
R2 es H;
R3 se selecciona entre el grupo que consiste en
fenilo, 2-fluorofenilo, 2-hidroxifenilo, 4-aminofenilo, 4-metoxifenilo, 4-nitrofenilo, 2-(ciclopropilcarbamoil)fenilo, 3- (ciclopropilcarbamoil)fenilo, 4-(ciclopropilcarbamoil)fenilo, 2,6-dimetilfenilo, 2-cloro-6-metilfenilo, 3,5- dimetoxifenilo, 3-ciano-5-metoxifenilo, 3-carbamoil-5-metoxifenilo, 4-cloro-3-fluorofenilo, 2,3-diclorofenilo, 4-cloro- 3-(trifluorometil)fenilo,…
Fragmento Fc de IgG4 que comprende una región bisagra modificada.
Secciones de la CIP Química y metalurgia Necesidades corrientes de la vida
(10/10/2018). Inventor/es: JUNG, SUNG YOUB, LEE,JONG-SOO, CHOI,IN-YOUNG, PARK,SUNG HEE, HUH,YONG HO. Clasificación: C12N15/13, A61K47/42, C12N15/63, C07K7/08.
Un fragmento Fc de IgG4 modificado que comprende una región bisagra modificada, en el que la región bisagra representada por la siguiente secuencia de aminoácidos se modificó delecionando parte de la secuencia de aminoácidos para incluir solo un resto de cisteína:
Glu-Ser-Lys-Tyr-Gly-Pro-Pro-Cys-Pro-Ser-Cys-Pro.
PDF original: ES-2685617_T3.pdf
Formulación farmacéutica estable para administración oral, que comprende levocetirizina o una sal farmacéuticamente aceptable de la misma y montelukast o una sal farmacéuticamente aceptable del mismo.
Sección de la CIP Necesidades corrientes de la vida
(11/04/2018). Inventor/es: WOO, JONG SOO, PARK,JAE HYUN, KIM,KYEONG SOO, KIM,YONG IL, KWON,TAEK KWAN, KIM,DONG HO. Clasificación: A61K47/12, A61K31/495, A61K9/20, A61K9/48, A61K9/14, A61K31/496, A61K9/28, A61K31/4704, A61K31/4965.
Una formulación farmacéutica para administración oral para prevenir o tratar la rinitis alérgica o el asma, que comprende:
(a) una primera porción de partículas que comprende levocetirizina o una sal farmacéuticamente aceptable de la misma y un ácido orgánico; y
(b) una segunda porción de partículas que comprende montelukast o una sal farmacéuticamente aceptable del mismo,
en la que dichas primera o segunda porciones de partículas están en forma de minicomprimido; y dicho ácido orgánico se selecciona entre el grupo que consiste en ácido cítrico, ácido tartárico, ácido succínico, ácido ascórbico, y una mezcla de los mismos.
PDF original: ES-2671428_T3.pdf
Composición farmacéutica que comprende un derivado de amida que inhibe el crecimiento de las células cancerosas y un lubricante de una sal no metálica.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(15/03/2017). Inventor/es: WOO, JONG SOO, PARK,JAE HYUN, KIM,KYEONG SOO, KIM,YONG IL, KIM,YO HAN, KIM,JIN CHEUL. Clasificación: A61K9/20, C07D401/12, A61K31/517.
Una composición farmacéutica que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo y un lubricante seleccionado de lubricante no metálico, donde dicho lubricante de sal no metálica se selecciona del grupo que consiste de ésteres de ácido graso, ácidos grasos, aceites, polietilenglicoles (PEGs), almidón, talco, y una mezcla de los mismos**Fórmula**.
PDF original: ES-2625268_T3.pdf
Método para preparar 1-(4-(4-(3,4-dicloro-2-fluorofenilamino)-7-metoxiquinazolin-6-iloxi)piperidin-1-il)prop-2-en-1-ona.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(06/07/2016). Inventor/es: MOON, YOUNG, HO, BANG,KEUK CHAN, JUNG,JAE HYUK. Clasificación: A61P35/00, C07D401/12, A61K31/517.
Un método para preparar el compuesto de fórmula (I), que comprende la etapa de permitir que el compuesto de fórmula (II) reaccione con el compuesto de fórmula (III) en un disolvente aprótico polar inerte en presencia de una base:**Fórmula**
en donde X es tosiloxi (OT), mesiloxi (OMs), trifluorometanosulfonato, fluorosulfonato o halógeno; y Y es etenilo o halogenoetilo.
PDF original: ES-2662863_T3.pdf
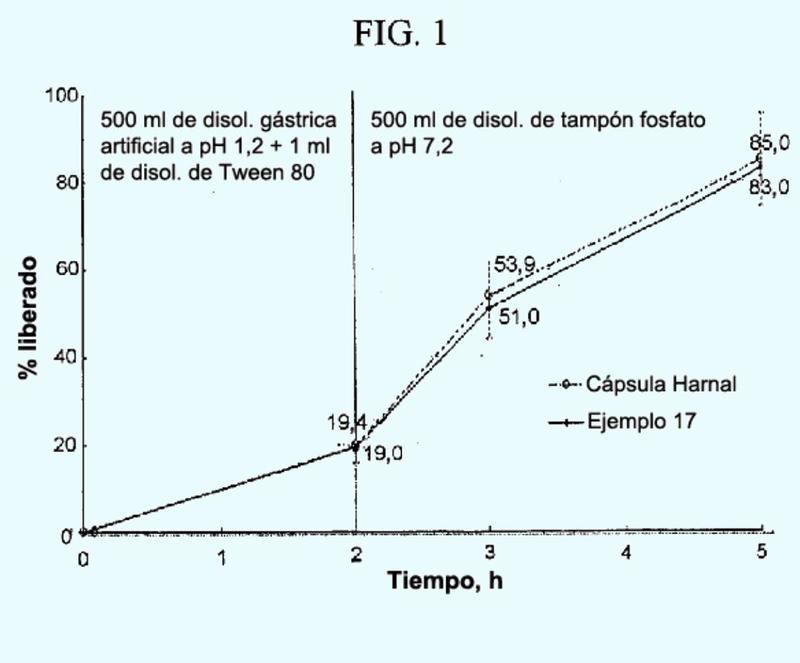
Composición para administración oral de hidrocloruro de tamsulosina y formulación granulada de liberación controlada que comprende la misma.
Sección de la CIP Necesidades corrientes de la vida
(13/01/2016). Ver ilustración. Inventor/es: WOO, JONG SOO, CHANG, HEE-CHUL. Clasificación: A61K9/16, A61K31/18, A61K47/38.
Composición para administración oral de hidrocloruro de tamsulosina que comprende hidrocloruro de tamsulosina, poli(acetato de vinilo) y una hidroxipropilmetilcelulosa soluble en agua, en la que la cantidad de poli(acetato de vinilo) oscila entre 20 y 1000 partes en peso basándose en 1 parte en peso de hidrocloruro de tamsulosina.
PDF original: ES-2561940_T3.pdf
Formulación de liberación controlada para la administración oral de metformina.
(12/08/2015) Formulación de liberación controlada para administración oral de metformina o una sal farmacéuticamente aceptable de la misma que comprende metformina o una sal farmacéuticamente aceptable de la misma como principio activo; una combinación de un poli(óxido de etileno) y goma xantana como portador para liberación controlada; y un aditivo farmacéuticamente aceptable, en la que la razón en peso de metformina o una sal farmacéuticamente aceptable de la misma:portador oscila entre 1:0,01 y 1:1.
Composición de un complejo oral que comprende un éster de ácidos grasos omega-3 y un inhibidor de HMG-CoA reductasa.
Sección de la CIP Necesidades corrientes de la vida
(24/09/2014). Inventor/es: WOO, JONG SOO, PARK,JAE HYUN, KIM,YONG IL, YOON,EUN JIN, IM,HO TAEK, SHIN,YOON SUB. Clasificación: A61K47/48, A61K9/48, A61K31/20, A61K9/28.
Composición de un complejo oral, que comprende:
(a) un núcleo de cápsula blanda que comprende ésteres de ácidos grasos omega-3;
(b) una primera capa de recubrimiento que envuelve el núcleo de cápsula blanda y comprende un material de recubrimiento hidrofóbico; y
(c) una segunda capa de recubrimiento depositada sobre la primera capa de recubrimiento, que comprende:
(i) rosuvastatina o una sal farmacéuticamente aceptable de la misma, y
(ii) alcohol polivinílico, alcohol polivinílico-copolímero de injerto de polietilenglicol o una mezcla de los mismos.
PDF original: ES-2550392_T3.pdf
Derivados de quinazolina como inhibidor múltiplex y método para la preparación de los mismos.
(17/06/2013) Compuesto de fórmula (I) o sal farmacéuticamente aceptable del mismo:
en el que
R1, R2, R3, R4 y R5 son cada uno independientemente hidrógeno, hidroxilo, halógeno, trifluorometilo, alquiloC1-C6, alcoxilo C1-C6, cicloalquilo C3-C7, hidroxi-alquilo C1-C5, alcoxi C1-C6-alquilo C1-C6, amino, aminoalquiloC1-C4, alquil C1-C6-amino, alcoxi C1-C6-carbonilo, alcoxi C1-C6-aminocarbonilo, aril-alcoxilo C1-C6,heteroaril-alcoxilo C1-C6 o arilo;
R6 es hidrógeno, alquilo C1-C6 o di(alquil C1-C6)-amino-alquilo C1-C6;
X es alquenil C2-C6-carbonilo o alquinil C2-C6-carbonilo opcionalmente sustituidos con R11, siendo R11halógeno, hidroxilo, amino, tiol, carbamoílo, trifluorometilo,…
Composición de microemulsión oral que comprende bifenildicarboxilato de dimetilo y silibina.
(03/06/2013) Composición de microemulsión oral para tratar una enfermedad hepática, que comprende
bifenildicarboxilato de dimetilo (BDD) y
silibina o un derivado de la misma, o un extracto de Carduus marianus que contiene silibina y derivadosde la misma, como principios activos; un co-tensioactivo; un tensioactivo; y un aceite, en la que elderivado de silibina es silicristina, silidiamina o isosilibina.
Procedimiento para la preparación de ácido Montelukast en medio iónico líquido.
(21/03/2013) Procedimiento para la preparación de ácido Montelukast de fórmula 1 o la sal sódica del mismo, comprendiendo laetapa de acoplamiento de un compuesto tiol de fórmula 2 con intermediario de Montelukast de fórmula 3 enpresencia de una base en un medio que comprende un compuesto iónico líquido seleccionado del grupo queconsiste en los compuestos de fórmulas 4a a 4e: **Fórmula**
Procedimiento para preparar voriconazol.
(08/03/2013) Un procedimiento para preparar voriconazol de formula (I) que comprende las etapas de:
a) someter el compuesto de fórmula (IV) a una reacción de acoplamiento de tipo Reformatsky con un compuesto defórmula (V) para obtener un compuesto de fórmula (III) que es un par de enantiómeros (2R,3S)/(2S,3R);
b) eliminar el derivado de tiol del compuesto del fórmula (III) para obtener el voriconazol racémico de fórmula (II); y
c) aislar el compuesto de formula (II) mediante resolución óptica usando un ácido ópticamente activo:en las que,
R es un alquilo C1-C4, benzotiazolilo, benzoxazolilo, imidazolilo, 1-metilimidazolilo, tiazolilo, piridilo,…
INHIBIDOR DE LA GLICOPROTEINA P, METODO PARA SU PREPARACION Y COMPOSICION FARMACEUTICA QUE COMPRENDE DICHO INHIBIDOR.
(05/07/2010) Un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo:
MONOHIDRATO DE L-MALATO DE AZITROMICINA CRISTALINO Y COMPOSICION FARMACEUTICA QUE LO CONTIENE.
(24/02/2010) Un monohidrato de L-malato de azitromicina cristalino de la fórmula (I): **(Ver fórmula)**
{kind=link}