Composición de liberación controlada de fármaco resistente a la tensión mecánica in vivo.
Matriz de liberación controlada de fármaco que consiste en uno o más fármacos, behenato de glicerilo y un éter de celulosa.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/EP2004/050814.
Solicitante: Aptalis Pharma Limited.
Nacionalidad solicitante: Irlanda.
Dirección: The Yard House, Killruddery Estate, Southern Cross Road Bray, County Wicklow IRLANDA.
Inventor/es: DE LUIGI BRUSCHI, STEFANO, PENGO, SERGIO.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A61K9/14 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 9/00 Preparaciones medicinales caracterizadas por un aspecto particular. › en estado especial, p. ej. polvos (microcápsulas A61K 9/50).
- A61K9/20 A61K 9/00 […] › Píldoras, pastillas o comprimidos.
Fragmento de la descripción:
Composición de liberación controlada de fármaco resistente a la tensión mecánica in vivo.
Campo de la invención
La presente invención se refiere a una composición farmacéutica de liberación controlada de fármaco caracterizada por una alta resistencia a la tensión mecánica in vivo y un perfil de absorción en el tiempo amplio y regular in vivo.
Estado de la técnica
Se usan ampliamente matrices hinchables para conseguir formas de dosificación monolíticas o multiparticuladas capaces de asegurar un perfil de liberación de fármaco según las necesidades terapéuticas. Se hace una mezcla dispersando el fármaco con polímeros hidrófilos solubles o insolubles más adyuvantes de compresión. Luego se granula la mezcla o se hace directamente comprimidos para conseguir la forma final de dosificación de liberación controlada. La liberación del fármaco sucede gracias a las propiedades de hinchamiento del polímero que constituye la matriz que se hidrata en presencia de medios acuosos que ejercen así el retraso de la liberación del fármaco.
Según la solubilidad del fármaco, el mecanismo de liberación se basa en la difusión a través de la matriz hinchada o en la erosión del polímero o en una combinación de las mismas.
La cinética de liberación de fármaco está gobernada por varios factores, es decir, solubilidad del fármaco, velocidad de hidratación del polímero, viscosidad y peso del polímero, tipo y cantidad de cargas, etc.
Una descripción exhaustiva de estos sistemas de liberación controlada se puede encontrar en la patente de EE.UU. 4.259.314 y en la patente de EE.UU. 4.680.323.
Los sistemas de matrices descritos en estas patentes están específicamente diseñados para asegurar una velocidad de disolución del fármaco in vitro que origine los niveles pico de fármaco en plasma esperados después de la toma.
Los expertos en la materia saben bien que la elección del tipo y cantidad de medio de disolución así como las condiciones de agitación adoptadas dependerán de la solubilidad del fármaco y de la ventana de absorción.
En algunos casos el procedimiento de disolución adoptado se puede usar para determinar una correlación in vivo-in vitro (IVIVC). Como resultado, conociendo la farmacocinética del fármaco, se pueden predecir niveles pico en plasma por los datos de velocidad de disolución del fármaco por medio de procedimientos matemáticos de convolución.
Sobre la base de la suposición de que la liberación de fármaco desde una forma de dosificación de liberación controlada es la etapa que limita la velocidad en el proceso de absorción, el perfil de absorción en el tiempo que resulta de la convolución matemática se puede considerar que es indicativo de una disolución in vivo (D. Young y col., In vitro-in vivo correlations
advances in experimetal medicine and biology, vol. 423 Plenum Press, ©1997 Nueva York y Londres).
Después de la toma, la capacidad de la forma de dosificación fabricada por un sistema de matriz hidrófila para soportar el peristaltismo in vivo, manteniendo así sus propiedades de liberación controlada a lo largo del tracto intestinal, es por lo tanto esencial para asegurar los niveles pico en plasma esperados.
Desafortunadamente, la escasa resistencia mecánica in vivo conferida al estado hinchado por los sistemas de matrices hidrófilas conocidos puede ser la causa de fallo de distribución farmacocinética incluso cuando la velocidad de disolución del fármaco in vitro cumple con las especificaciones definidas.
De hecho, la metodología basada en la determinación de la velocidad de disolución del fármaco como herramienta predictiva para estimar los niveles pico en plasma in vivo denota límites severos puesto que los aparatos de disolución comunes no someten mecánicamente a tensión a la forma de dosificación que mantiene una forma bien definida durante la prueba de disolución, por el contrario, el perfil de absorción in vivo generalmente muestra un nivel pico en plasma drástico, debido a la contusión mecánica de la forma de dosificación. Esta concentración alta de fármaco puede conducir a efectos fisiológicos indeseables.
Para conseguir los niveles pico en plasma esperados, llega a ser de suma importancia proporcionar a la forma de dosificación gelificable propiedades mecánicas adecuadas para que resista el peristaltismo in vivo sin que se afecten las propiedades de disolución que conducen a la velocidad de absorción in vivo deseada.
Del documento EP 0441245 se sabe incluir aceites endurecidos (es decir, sebo de vacuno endurecido, aceite de semilla de colza endurecido) en una forma de dosificación para aumentar su resistencia mecánica. Sin embargo, la inclusión de aquellos da como resultado un retraso general de la velocidad de liberación, volviendo así al fármaco menos biodisponible, especialmente en los momentos tempranos después de la administración.
El documento FR-A-2656525 describe una composición de liberación controlada de fármaco que comprende hidroxipropilmetilcelulosa y al menos un éster de glicerilo. El documento FR-A-2775597 describe composiciones farmacéuticas con biodisponibilidad de fármaco mejorada que comprenden ésteres de glicerilo. El documento EP-A-1262198 describe una composición de liberación retardada que comprende levodopa, carbidopa y al menos un éter de celulosa.
Todas las matrices de liberación controlada conocidas de la técnica anterior no son completamente satisfactorias para conseguir resistencia mecánica in vivo. Por lo tanto se siente la necesidad de formas de liberación controlada mejoradas que sean mecánicamente resistentes frente al peristaltismo in vivo y que ofrezcan una velocidad de liberación deseada in vivo; en particular, se siente la necesidad de conseguir una resistencia mecánica mejorada in vivo sin incurrir en el inconveniente de tiempos de liberación muy prolongados, manteniendo así un rápido comienzo de la acción inmediatamente después de la administración, y liberando la dosis entera de fármaco en tiempos aceptables.
Resumen
La presente solicitud se refiere a una matriz de liberación controlada de fármaco que consiste en behenato de glicerilo, un éter de celulosa y uno o más fármacos en relaciones específicas de peso.
El éter de celulosa es preferiblemente hidroxipropilmetilcelulosa, metilcelulosa, hidroxipropilcelulosa, hidroxietilcelulosa, acetato de celulosa, sus derivados o mezcla de los mismos.
Preferiblemente, el éter se caracteriza por una viscosidad aparente que varía en el intervalo de 15 cP a 100.000 cP (solución acuosa al 2% peso/volumen, 20ºC).
Asimismo, la presente invención se refiere a una composición farmacéutica de liberación controlada de fármaco en la que se mezcla dicha matriz con excipientes farmacéuticamente aceptables y se formula en una forma administrable oralmente caracterizada por una resistencia mecánica en el estado hinchado mayor que la misma composición sin éster de glicerilo. Esta resistencia mecánica conduce a una mejor predicción de la concentración de fármaco en plasma in vivo sobre la base de los estudios de cinética de liberación in vitro. Se pueden obtener formas de dosificación administrables oralmente por procedimientos conocidos por sí mismos: por ejemplo, se puede comprimir o granular directamente una mezcla de éter de celulosa, éster de glicerilo y uno o más fármacos, hacer después comprimidos, etc.
Finalmente, la presente invención se refiere a las explotaciones farmacológicas de la composición descrita.
Descripción de las figuras
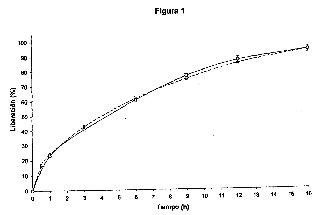
Figura 1
Velocidad de disolución de ISO-5-MN por comprimido de liberación controlada (100 mg/comprimido)
Cantidad en porcentaje (%) que se libera in vitro, (eje vertical) durante el tiempo (horas, eje horizontal) del comprimido de referencia G9A623 (-o-) y del comprimido I9A010 (-Δ-) según la presente invención. Las pruebas se hacen según el aparato 2 de disolución USP XXV, 500 ml, pH 1,2, 75 rpm, n = 6 comprimidos.
Figura 2
Concentración en plasma (situación alimentada) de comprimido de ISO-5-MN de liberación controlada (100 mg/ comprimido)
Concentración en plasma in vivo, (ng/ml,...
Reivindicaciones:
1. Matriz de liberación controlada de fármaco que consiste en uno o más fármacos, behenato de glicerilo y un éter de celulosa.
2. Matriz de liberación controlada de fármaco según la reivindicación 1, en la que el fármaco está presente en una cantidad de aproximadamente 20-95%, el behenato de glicerilo en una cantidad de aproximadamente 1-25%, y el éter de celulosa en una cantidad de aproximadamente 1-65%, en peso de dicha matriz.
3. Matriz de liberación controlada de fármaco según la reivindicación 2, en la que el fármaco está presente en una cantidad de aproximadamente 20-70%, el behenato de glicerilo en una cantidad de aproximadamente 1-25%, y el éter de celulosa en una cantidad de aproximadamente 1-65%, en peso de dicha matriz.
4. Matriz de liberación controlada de fármaco según la reivindicación 3, en la que el fármaco está presente en una cantidad de aproximadamente 20-30%, el behenato de glicerilo en una cantidad de aproximadamente 15-25%, y el éter de celulosa en una cantidad de aproximadamente 45-65%, en peso de dicha matriz.
5. Matriz de liberación controlada de fármaco según la reivindicación 4, en la que la cantidad de fármaco es aproximadamente 25%, el behenato de glicerilo es aproximadamente 20%, y el éter de celulosa es aproximadamente 55%, en peso de dicha matriz.
6. Matriz de liberación controlada de fármaco según la reivindicación 2, en la que el fármaco está presente en una cantidad de aproximadamente 70-95%, en peso de la matriz, el behenato de glicerilo está presente en una cantidad de aproximadamente 1-15%, en peso de la matriz, y el éter de celulosa está presente en una cantidad de aproximadamente 1-15%.
7. Matriz de liberación controlada de fármaco según la reivindicación 6, en la que el fármaco es carbidopa y/o levodopa.
8. Matriz de liberación controlada de fármaco según la reivindicación 7, en la que la relación en peso de behenato de glicerilo a éter de celulosa oscila desde aproximadamente 4:1 hasta 1:1.
9. Matriz de liberación controlada de fármaco según las reivindicaciones 1-8, en la que el éter de celulosa se elige entre hidroxipropilmetilcelulosa, metilcelulosa, hidroxipropilcelulosa, hidroxietilcelulosa, acetato de celulosa, sus derivados o mezcla de los mismos.
10. Matriz de liberación controlada de fármaco según las reivindicaciones 1-9, en la que el éter de celulosa se caracteriza por una viscosidad aparente que varía en el intervalo de 15 cP a 100.000 cP (solución acuosa al 2% peso/volumen, 20ºC).
11. Composición farmacéutica de liberación controlada de fármaco que comprende la matriz que se describe en las reivindicaciones 1-10, asociada con excipientes farmacéuticamente aceptables y formulada en una forma administrable por vía oral.
12. Composición farmacéutica de liberación controlada de fármaco según la reivindicación 11, en la que la matriz representa al menos el 40% en peso de dicha composición farmacéutica.
13. Composición farmacéutica de liberación controlada de fármaco según la reivindicación 11, en la que la matriz representa al menos el 60% en peso de dicha composición farmacéutica.
14. Composición farmacéutica de liberación controlada de fármaco según las reivindicaciones 11-13, en la que los excipientes se seleccionan entre deslizantes, aglutinantes, sabores, conservantes, agentes tamponadores, agentes colorantes, cargas, lubricantes.
15. Composición farmacéutica de liberación controlada de fármaco según las reivindicaciones 11-14, en la que la forma administrable por vía oral es un comprimido, minicomprimido o granulado.
16. Procedimiento para preparar una composición de liberación controlada, que comprende mezclar conjuntamente un fármaco, un behenato de glicerilo y un éter de celulosa.
17. Procedimiento según la reivindicación 16, en el que el éter de celulosa se elige entre hidroxipropilmetilcelulosa, metilcelulosa, hidroxipropilcelulosa, hidroxietilcelulosa, acetato de celulosa, sus derivados o mezcla de los mismos.
18. Procedimiento según las reivindicaciones 16-17, en el que el éter de celulosa se caracteriza por una viscosidad aparente que varía en el intervalo de 15 cP a 100.000 cP (solución acuosa al 2% peso/volumen, 20ºC).
19. Procedimiento según las reivindicaciones 16-18, en el que dicho fármaco, behenato de glicerilo y éter de celulosa se mezclan con excipientes farmacéuticamente aceptables y la mezcla final resultante se formula en una forma administrable por vía oral.
20. Procedimiento según las reivindicaciones 16-19, en el que la suma de dicho fármaco, behenato de glicerilo y éter de celulosa representa al menos el 40% en peso de dicha mezcla final.
21. Procedimiento según las reivindicaciones 16-20, en el que la suma de dicho fármaco, behenato de glicerilo y éter de celulosa representa al menos el 60% en peso de dicha mezcla final.
22. Procedimiento según las reivindicaciones 16-21, en el que los excipientes se seleccionan entre deslizantes, aglutinantes, sabores, conservantes, agentes tamponadores, agentes colorantes, cargas, lubricantes.
23. Procedimiento según las reivindicaciones 16-22, en el que la forma administrable por vía oral es un comprimido, minicomprimido o granulado.
Patentes similares o relacionadas:
Preparación sólida que contiene colorante, del 29 de Julio de 2020, de DAIICHI SANKYO COMPANY, LIMITED: Preparación farmacéutica sólida que comprende monobencenosulfonato de ácido [(1R,5S,6S)-6-(aminometil)-3- etilbiciclo[3.2.0]hept-3-en-6-il]acético […]
Formulación de vitamina D de liberación modificada estabilizada y método de administración de la misma, del 22 de Julio de 2020, de EirGen Pharma Ltd: Una formulacion oral de liberacion controlada de un compuesto de vitamina D que comprende uno o ambos de 25- hidroxivitamina D2 y 25-hidroxivitamina D3, la formulacion […]
Métodos y composiciones para la administración oral de proteínas, del 22 de Julio de 2020, de Entera Bio Ltd: Una única composición farmacéutica oral que comprende una proteína que tiene un peso molecular de hasta 100.000 Daltons, siendo dicha proteína PTH; […]
Macrogols para aplicación a la mucosa, y sus usos terapéuticos, del 15 de Julio de 2020, de S.I.I.T. S.R.L.-SERVIZIO INTERNAZIONALE IMBALLAGGI TERMOSALDANTI: Composición farmacéutica en forma sólida que comprende, por unidad de dosificación, entre 5 y 400 mg de un PEG con un grado de 3000 o más, para uso en el tratamiento […]
Composición farmacéutica que comprende un agente antipsicótico atípico y método para su preparación, del 15 de Julio de 2020, de PHARMATHEN S.A.: Comprimido de liberación controlada de Paliperidona en forma de comprimido de varias capas que comprende: a) un núcleo de matriz que comprende […]
Composiciones y métodos para tratar el virus de la hepatitis C, del 15 de Julio de 2020, de Gilead Pharmasset LLC: Una composición farmacéutica que comprende: a) de aproximadamente el 25% a aproximadamente el 35% p/p de GS-7977 cristalino que tiene la estructura **(Ver […]
Preparación para el control del peso corporal a base de quitosano y celulosa, del 1 de Julio de 2020, de S.I.I.T. S.R.L.-SERVIZIO INTERNAZIONALE IMBALLAGGI TERMOSALDANTI: Una composición oral sólida que contiene una combinación de quitosano de hongos o levaduras, celulosa amorfa en polvo y opcionalmente excipientes.
Granulados secos de polvos de sílice mesoporosa, del 1 de Julio de 2020, de FORMAC PHARMACEUTICALS N.V: Un granulado seco que comprende desde el 50% al 100% p/p de sílice mesoporosa ordenada que tiene una organización bidimensional hexagonalmente […]