MÉTODO DE ANÁLISIS DE ÁCIDO NUCLEICO PARA ANALIZAR EL PATRÓN DE METILACIÓN DE ISLAS CPG EN DIFERENTES MUESTRAS.
Método para determinar la metilación de un ácido nucleico que comprende las siguientes etapas:
a) fragmentación de una muestra de ADN genómico con al menos un enzima de restricción sensible a metilación y al menos un enzima de restricción no sensible a metilación, b) ligación, en los extremos de los fragmentos de ADN obtenidos, de adaptadores específicos donde uno de los adaptadores específicos comprende una secuencia promotora funcional, c) amplificación de los fragmentos que incluyen ambos adaptadores utilizando cebadores específicos basados en los adaptadores, d) marcaje de los fragmentos de ADN amplificados mediante transcripción in vitro con una ARN polimerasa capaz de iniciar la transcripción a partir de la secuencia promotora contenida en uno de los adaptadores utilizando una mezcla de nucleótidos y e) determinación del estado de metilación de la muestra
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/EP2008/053748.
Solicitante: ORYZON GENOMICS, S.A..
Nacionalidad solicitante: España.
Inventor/es: MAES,TAMARA, AIBAR DURAN,ELENA, DURANY TURK,Olga.
Fecha de Publicación: .
Fecha Solicitud PCT: 28 de Marzo de 2008.
Clasificación Internacional de Patentes:
- C12Q1/68D2E1
Clasificación PCT:
- C12Q1/68 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12Q PROCESOS DE MEDIDA, INVESTIGACION O ANALISIS EN LOS QUE INTERVIENEN ENZIMAS, ÁCIDOS NUCLEICOS O MICROORGANISMOS (ensayos inmunológicos G01N 33/53 ); COMPOSICIONES O PAPELES REACTIVOS PARA ESTE FIN; PROCESOS PARA PREPARAR ESTAS COMPOSICIONES; PROCESOS DE CONTROL SENSIBLES A LAS CONDICIONES DEL MEDIO EN LOS PROCESOS MICROBIOLOGICOS O ENZIMOLOGICOS. › C12Q 1/00 Procesos de medida, investigación o análisis en los que intervienen enzimas, ácidos nucleicos o microorganismos (aparatos de medida, investigación o análisis con medios de medida o detección de las condiciones del medio, p. ej. contadores de colonias, C12M 1/34 ); Composiciones para este fin; Procesos para preparar estas composiciones. › en los que intervienen ácidos nucleicos.
Países PCT: Austria, Bélgica, Suiza, Alemania, Dinamarca, España, Francia, Reino Unido, Grecia, Italia, Liechtensein, Luxemburgo, Países Bajos, Suecia, Mónaco, Portugal, Irlanda, Eslovenia, Finlandia, Rumania, Chipre, Lituania, Letonia.
PDF original: ES-2374194_T3.pdf
Fragmento de la descripción:
Método de análisis de ácido nucleico para analizar el patrón de metilación de islas CpG en diferentes muestras.
CAMPO DE LA INVENCIÓN
La presente invención se enmarca en el campo de la biología molecular. En particular, la presente invención tiene por objeto un método de análisis de ácido nucleico que puede ser utilizado para analizar el patrón de metilación que presentan las islas CpG en diferentes muestras.
ANTECEDENTES DE LA INVENCIÓN
En los últimos tiempos se ha puesto de manifiesto que los factores epigenéticos pueden jugar un papel muy importante en el control genético de procesos celulares, entre ellos el desarrollo del cáncer. Entre estos factores epigenéticos, la metilación de determinados fragmentos de ADN es uno de los más relevantes.
La metilación del ADN es un proceso epigenético que participa en la regulación de la expresión génica de dos maneras: directamente al impedir la unión de factores de transcripción, e indirectamente propiciando la estructura "cerrada" de la cromatina (Singal R, & Ginder GD. DNA methylation. Blood. 1999 Jun 15;93 (12) :4059-70) . El ADN presenta regiones de 1000-1500 pb ricas en dinucleótidos CpG ("islas CpG") , que son reconocidas por las enzimas ADN-metiltransferasas, las cuales, durante la replicación del ADN metilan el carbono de la posición 5 de las citosinas de la cadena recién sintetizada, manteniéndose así la memoria del estado metilado en la molécula hija de ADN. En general se considera que la metilación es un proceso unidireccional, de manera que, cuando una secuencia CpG se metila de novo, esta modificación se hace estable y es heredada como un patrón de metilación clonal. Por otra parte, la alteración en el estado de metilación de genes reguladores (hipometilación o hipermetilación) , como evento primario, se asocia frecuentemente con el proceso neoplásico y es proporcional a la severidad de la enfermedad (Paluszczak J, & Baer-Dubowska W. Epigenetic diagnostics of cancer - the application of DNA methylation markers. J Appl Genet. 2006;47 (4) :365-75) .
Los genomas de las células preneoplásicas, cancerosas y envejecidas comparten tres cambios importantes en los niveles de metilación, como eventos tempranos en el desarrollo de algunos tumores. Primero, la hipometilación de la heterocromatina que conduce a una inestabilidad genómica e incrementa los eventos de recombinación mitótica; segundo, hipermetilación de genes individuales y, finalmente hipermetilación de la islas CpG de genes constitutivos y genes supresores de tumor. Los dos niveles de metilación pueden presentarse de manera individual o simultánea; en general, la hipermetilación está involucrada en el silenciamiento de genes y la hipometilación en la sobreexpresión de ciertas proteínas involucradas en los procesos de invasión y metástasis.
Las estrategias metodológicas para el análisis del estado de metilación de las islas CpG han estado en constante evolución. La mayoría de los métodos se basan en la transformación química de las citosinas no metiladas a uracilos, por el tratamiento con bisulfito de sodio, que no afecta las 5-metilcitosinas, y marca de forma fidedigna e individual el estado metilado o no metilado de los dinucleótidos CpG. La modificación del ADN, su amplificación por la reacción en cadena de la polimerasa (PCR) y/o secuenciación automatizada son las técnicas más usadas con este propósito (Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol. 2005;45:629-56) .
En los últimos años la tecnología basada en el análisis del ADN metilado es considerada una poderosa herramienta para el diagnóstico, terapia y pronóstico de enfermedad, así como en el campo de la medicina forense, farmacogenética y en estudios epidemiológicos. La asociación entre el estado hipometilado del ADN y cáncer, y posteriormente su relación con la hipermetilación, se conocen desde 1983; sin embargo, en los últimos cinco años, por el impulso de las nuevas estrategias moleculares para el estudio de la metilación de novo de las islas CpG, el análisis del ADN metilado se ha convertido en un poderoso biomarcador para la detección temprana de cáncer; además, permite clasificar los cánceres considerando los subtipos histológicos, el grado de malignidad, diferencias en la respuesta al tratamiento, y los diversos pronósticos. Una importante y reciente aplicación es precisamente su uso como biomonitor de respuesta a terapia y predictor del pronóstico del cáncer.
La metilación del ADN es un marcador epigenético del silenciamiento de genes con aplicación en diversos campos de investigación de la genética y la biomedicina, que mediante la aplicación de procesos metodológicos moleculares permite la distinción individual de los patrones de metilación de las islas CpG. Por otra parte, las características de metilación de los genes involucrados en la neoplasia permiten la clasificación y el pronóstico de los cánceres, y el seguimiento del tratamiento.
El desarrollo de las micromatrices de ADN (también denominadas chips o microarrays de ADN) , ha permitido que sean rápidamente integradas en los estudios genómicos, con lo que se obtienen unos mejores niveles de resolución y sensibilidad en el análisis comparativo de ADN genómico y una mayor capacidad de reproducción que permiten la detección fiable de alteraciones a nivel individual en los genes. Gracias a su versatilidad, la tecnología de micromatrices de ADN presenta aplicaciones en los campos de la transcriptómica, la genética y la epigenética.
Hatada et al., “A microarray-based method for detecting methylated loci”, Journal of Human Genetics-2002, vol. 47, p. 448-451, describía un método basado en un micromatriz denominado chip de amplificación metilación de ADN (MAD) que comprende las etapas de digerir el ADN con el enzima sensible a metilación SmaI y el enzima no sensible a metilación XmaI, ligar con un adaptador, amplificar por PCR, marcar Cy3/Cy5 y co-hibridar a un micromatriz. El método permite la amplificación específica de las islas CpG, lo que lleva a amplicones útiles en la hibridación que resultan en una elevada sensibilidad. Hatada et al., propone el uso del método para detectar genes o genes supresores de tumor cuyos niveles de expresión son demasiado bajos como para ser detectados mediante un método basado en expresión.
DESCRIPCIÓN RESUMIDA DE LA INVENCIÓN
La presente invención se refiere a un método de análisis de ácido nucleico que comprende ADN. La presente invención se refiere a un método de análisis de ácido nucleido que comprende una fragmentación del ADN, la ligación de adaptadores específicos, una etapa de amplificación y el marcaje de las muestras utilizando ARN polimerasa. En esta etapa se generan un conjunto de fragmentos de ARN representativos de los fragmentos de ADN a analizar. Estos fragmentos de ARN pueden, por ejemplo, hibridarse posteriormente con una micromatriz de ADN para realizar el análisis. El método de la presente invención puede utilizarse para identificar selectivamente eventos de metilación en las muestras analizadas.
En una realización, la invención proporciona un método para determinar la metilación de un ácido nucleico. El método de esta realización comprende proporcionar u obtener una muestra que tiene un ácido nucleico, tratar la muestra de ácido nucleico con un enzima de restricción no sensible a metilación, tratar la muestra con un enzima de restricción sensible a metilación, ligar un adaptador al sitio creado por el enzima de restricción no sensible a metilación, someter los ácidos nucleicos ligados al adaptador a condiciones de amplificación, marcar el ácido nucleico amplificado mediante transcripción in vitro, y detectar el ácido nucleico marcado.
En otra realización, la invención proporciona un método para determinar la metilación de un ácido nucleico. El método de esta realización comprende (1) proporcionar u obtener una muestra que tiene ácido nucleico, (2) tratar la muestra de ácido nucleico con un enzima de restricción no sensible a metilación, (3) tratar la muestra con un enzima de restricción sensible a metilación, (4) ligar un adaptador al sitio creado por el enzima de restricción no sensible a metilación, (5) ligar un adaptador al sitio creado por el enzima de restricción sensible a metilación, en donde dicho adaptador está manipulado genéticamente para incluir un promotor adecuado para la transcripción... [Seguir leyendo]
Reivindicaciones:
1. Método para determinar la metilación de un ácido nucleico que comprende las siguientes etapas:
a) fragmentación de una muestra de ADN genómico con al menos un enzima de restricción sensible a metilación y al menos un enzima de restricción no sensible a metilación, b) ligación, en los extremos de los fragmentos de ADN obtenidos, de adaptadores específicos donde uno de los adaptadores específicos comprende una secuencia promotora funcional, c) amplificación de los fragmentos que incluyen ambos adaptadores utilizando cebadores específicos basados en los adaptadores, d) marcaje de los fragmentos de ADN amplificados mediante transcripción in vitro con una ARN polimerasa capaz de iniciar la transcripción a partir de la secuencia promotora contenida en uno de los adaptadores utilizando una mezcla de nucleótidos y e) determinación del estado de metilación de la muestra.
2. Método según la reivindicación 1, en donde la fragmentación de una muestra de ADN genómico se realiza mediante digestión en primer lugar con al menos un enzima de restricción no sensible a metilación y posteriormente con al menos un enzima de restricción sensible a metilación; o en donde la fragmentación de una muestra de ADN genómico se realiza mediante digestión en primer lugar con al menos un enzima de restricción sensible a metilación y posteriormente con al menos un enzima de restricción no sensible a metilación; o en donde la fragmentación de una muestra de ADN genómico se realiza mediante digestión con al menos un enzima de restricción no sensible a metilación y simultáneamente con al menos un enzima de restricción sensible a metilación.
3. Método según la reivindicación 1, en donde el enzima de restricción no sensible a metilación reconoce una diana de de enzimas de restricción de 4, 5 ó 6 pares de bases.
4. Método según la reivindicación 3, en donde el enzima de restricción no sensible a metilación se selecciona del grupo que comprende BfaI, TaqI, MseI y NdeI.
5. Método según la reivindicación 1, en donde el enzima de restricción sensible a metilación reconoce una diana de enzimas de restricción de 4, 5 ó 6 pares de bases.
6. Método según la reivindicación 5, en donde el enzima de restricción sensible a metilación se selecciona del grupo que comprende SmaI, PauI, TspMI, BsePI y BssHII.
7. Método según la reivindicación 1, en donde el adaptador específico que comprende una secuencia promotora funcional es el adaptador específico para el enzima de restricción sensible a metilación.
8. Método según la reivindicación 1, en donde el marcaje comprende la incorporación de análogos de nucleótidos que contienen sustancias marcadoras detectables directamente, tales como fluoróforos, análogos de nucleótidos que incorporan sustancias marcadoras detectables en una reacción posterior, tales como biotina o haptenos, o cualquier otro tipo de sustancia marcadora de ácidos nucleicos.
9. Método según la reivindicación 1, en donde la ARN polimerasa se selecciona del grupo que comprende ARN polimerasa de T7, ARN polimerasa de T3 y ARN polimerasa de SP6.
10. Método según la reivindicación 1, en donde la determinación del estado de metilación de la muestra se realiza mediante hibridación de los fragmentos de ARN obtenidos en la etapa d) con los oligonucleótidos inmovilizados sobre una micromatriz de ADN, la detección del marcaje incorporado en los fragmentos a analizar y la comparación cuantitativa de los valores de las señales de los fragmentos hibridados con los valores de las señales de referencia.
11. Método según la reivindicación 10, en donde los oligonucleótidos inmovilizados sobre la micromatriz se diseñan de forma que incluyan la diana de restricción del enzima de restricción sensible a metilación; o en donde los oligonucleótidos inmovilizados sobre la micromatriz se diseñan de forma que estén localizados entre las dianas de restricción del enzima de restricción sensible a metilación.
12. Kit que comprende los reactivos, enzimas y aditivos apropiados para llevar a cabo el método de cualquiera de las reivindicaciones 1 a 11.
13. Uso del método reivindicado en cualquiera de las reivindicaciones 1 a 11 para analizar el patrón de metilación que presentan las islas CpG en la muestra analizada.
14. Uso del método reivindicado en cualquiera de las reivindicaciones 1 a 11 para el diagnóstico de un estado patológico.
15. Uso del método según la reivindicación 14, donde el estado patológico se selecciona del grupo que consiste en cáncer y una enfermedad neurodegenerativa.
Marcador Marcador ADN 1 2 3 4 5 6 7 8 9 10 11 ADN
11501 bp
2838 bp
2459 bp
1700 bp
805 bp
514 bp
264 bp
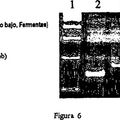
FIG. 2
Marcador Marcador ADN 1 2 3 45 6 7 8 Vacío ADN
11501 bp
2838 bp
2459 bp
1700 bp
805 bp
514 bp
264 bp
FIG. 3
Marcador Marcador ARN 1 2 Vacío ARN
6000 bp 4000 bp 3000 bp 2000 bp 1500 bp 1000 bp
500 bp
200 bp
FIG. 4
REFERENCIAS CITADAS EN LA DESCRIPCIÓN
Esta lista de referencias citadas por el solicitante es únicamente para la comodidad del lector. No forma parte del documento de la patente europea. A pesar del cuidado tenido en la recopilación de las referencias, no se pueden excluir errores u omisiones y la EPO niega toda responsabilidad en este sentido.
Documentos de patentes citados en la descripción
Literatura diferente de patentes citadas en la descripción
Patentes similares o relacionadas:
 PROCEDIMIENTO PARA LA CARACTERIZACIÓN DE POLINUCLEÓTIDOS, del 25 de Febrero de 2011, de LINGVITAE AS: Procedimiento para determinar la secuencia de un polinucleótido diana, que comprende las etapas siguientes: (i)tratar una muestra de un polinucleótido diana bicatenario […]
PROCEDIMIENTO PARA LA CARACTERIZACIÓN DE POLINUCLEÓTIDOS, del 25 de Febrero de 2011, de LINGVITAE AS: Procedimiento para determinar la secuencia de un polinucleótido diana, que comprende las etapas siguientes: (i)tratar una muestra de un polinucleótido diana bicatenario […]
Método para analizar ácido nucleico molde, método para analizar sustancia objetivo, kit de análisis para ácido nucleico molde o sustancia objetivo y analizador para ácido nucleico molde o sustancia objetivo, del 29 de Julio de 2020, de Kabushiki Kaisha DNAFORM: Un método para analizar un ácido nucleico molde, que comprende las etapas de: fraccionar una muestra que comprende un ácido nucleico molde […]
MÉTODOS PARA EL DIAGNÓSTICO DE ENFERMOS ATÓPICOS SENSIBLES A COMPONENTES ALERGÉNICOS DEL POLEN DE OLEA EUROPAEA (OLIVO), del 23 de Julio de 2020, de SERVICIO ANDALUZ DE SALUD: Biomarcadores y método para el diagnostico, estratificación, seguimiento y pronostico de la evolución de la enfermedad alérgica a polen del olivo, kit […]
Detección de interacciones proteína a proteína, del 15 de Julio de 2020, de THE GOVERNING COUNCIL OF THE UNIVERSITY OF TORONTO: Un método para medir cuantitativamente la fuerza y la afinidad de una interacción entre una primera proteína de membrana o parte de la misma y una […]
Secuenciación dirigida y filtrado de UID, del 15 de Julio de 2020, de F. HOFFMANN-LA ROCHE AG: Un procedimiento para generar una biblioteca de polinucleótidos que comprende: (a) generar una primera secuencia del complemento (CS) de un polinucleótido diana a partir […]
Métodos para la recopilación, estabilización y conservación de muestras, del 8 de Julio de 2020, de Drawbridge Health, Inc: Un método para estabilizar uno o más componentes biológicos de una muestra biológica de un sujeto, comprendiendo el método obtener un […]
Evento de maíz DP-004114-3 y métodos para la detección del mismo, del 1 de Julio de 2020, de PIONEER HI-BRED INTERNATIONAL, INC.: Un amplicón que consiste en la secuencia de ácido nucleico de la SEQ ID NO: 32 o el complemento de longitud completa del mismo.
Composiciones para modular la expresión de SOD-1, del 24 de Junio de 2020, de Biogen MA Inc: Un compuesto antisentido según la siguiente fórmula: mCes Aeo Ges Geo Aes Tds Ads mCds Ads Tds Tds Tds mCds Tds Ads mCeo Aes Geo mCes Te (secuencia […]