METODOS CORRECTORES DE TRATAMIENTO DE RESULTADOS DE EXPERIMENTOS TRANSCRIPTOMICOS OBTENIDOS POR ANALISIS DIFERENCIAL.
Método corrector llevado a cabo por ordenador de tratamiento de resultados de experimentos transcriptómicos obtenidos por análisis diferencial,
consistente en las etapas siguientes:
- obtención de los resultados del nivel de expresión de genes en una condición de referencia y cálculo del nivel medio de expresión de cada uno de dichos genes;
- obtención de los resultados del nivel de expresión de dichos genes en una condición de tratamiento y cálculo del nivel medio de expresión para cada uno de dichos genes;
- cálculo del coeficiente de modulación del nivel de expresión para cada uno de dichos genes;
- cálculo de un p-value asociado a cada coeficiente de modulación;
- cálculo de curvas isobáricas de p-value en función del nivel medio de expresión de cada uno de dichos genes en la condición de referencia;
caracterizado por incluir además una etapa de cálculo y de asociación de un coeficiente de modulación mediano en la curva isobárica de cada p-value observado
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/FR2007/052562.
Solicitante: GALDERMA RESEARCH & DEVELOPMENT.
Nacionalidad solicitante: Francia.
Dirección: LES TEMPLIERS 2400 ROUTE DES COLLES,06410 BIOT.
Inventor/es: AUBERT, JEROME, FOGEL,PAUL, ZUGAJ,DIDIER, LE GOFF,JEAN-MARC, DERET,SOPHIE.
Fecha de Publicación: .
Fecha Concesión Europea: 31 de Marzo de 2010.
Clasificación Internacional de Patentes:
- G06F19/00C4
Clasificación PCT:
- G06F19/00
Fragmento de la descripción:
Métodos correctores de tratamiento de resultados de experimentos transcriptómicos obtenidos por análisis diferencial.
La presente invención se relaciona con métodos correctores de tratamiento de resultados de experimentos transcriptómicos obtenidos por análisis diferencial. Se relaciona más particularmente con el tratamiento de tales resultados en el caso de experimentos realizados sobre chips de ADN. Los experimentos transcriptómicos tienen por objeto la identificación de genes de interés o de grupos de genes de interés.
Generalmente, el nivel de expresión de estos genes de interés o de estos grupos de genes de interés varía significativamente, por ejemplo, en respuesta a una señal. Cuando se realiza el análisis de resultados de experimentos transcriptómicos, por ejemplo por medio de chips de ADN, es habitual seleccionar los genes que presentan la mayor modulación, es decir, la mayor variación de su nivel de expresión. El nivel de esta modulación, también llamado coeficiente de modulación, se define como la razón del nivel de expresión observado en un experimento, por ejemplo en una condición llamada de tratamiento, con respecto al observado en otro experimento, por ejemplo en una condición llamada de referencia.
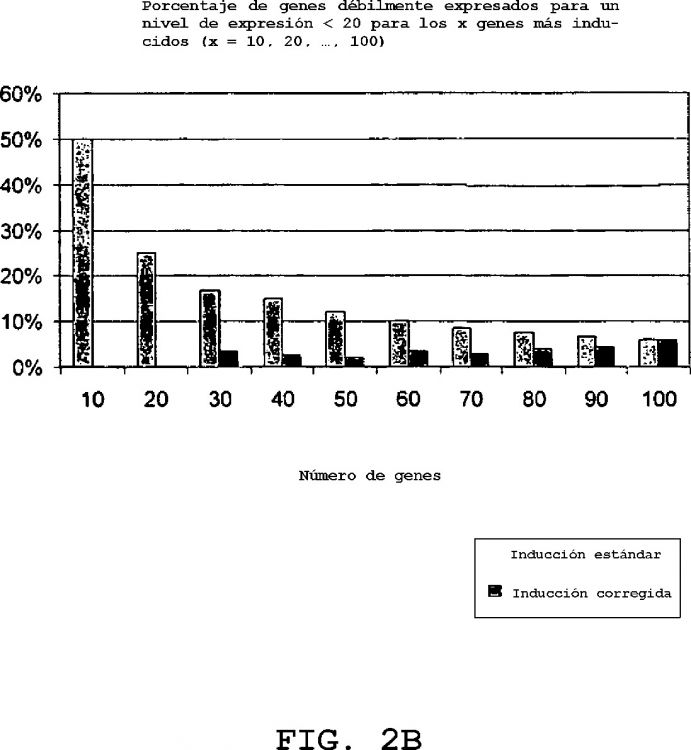
El examen de los resultados muestra que, cuanto más se utiliza un nivel de modulación elevado para restringir el número de genes seleccionados, más se favorece, en la selección realizada, la emergencia de genes cuyo nivel de expresión está, en la condición de referencia, próximo al límite de detección. Ahora bien, no existe ningún argumento biológico para explicar la razón por la cual los genes menos expresados en la condición de referencia serían los genes más intensamente modulados durante un tratamiento. Esta selección introduce, pues, un sesgo y lleva a ignorar genes que presentan un nivel de modulación menos elevado por el simple hecho de expresarse más en la condición de referencia.
Si se estima el coeficiente de modulación del nivel de expresión a partir de varias observaciones de un mismo gen sobre varios chips correspondientes a la misma condición, es decir, a partir de réplicas de la condición de referencia o de la condición de tratamiento, se demuestra que el coeficiente de modulación y el nivel medio de expresión de los genes evolucionan en sentido inverso [R. Mansourian et al., The Global error assessment (GEA) model for the selection of differentially expressed genes in microarray data, Bioinformatics Advance Access, 2004]. En otras palabras, cuanto más bajo es el nivel de expresión de un gen en varias réplicas de una condición de referencia, más elevado es su coeficiente de modulación en respuesta al tratamiento, calculado a partir de varias réplicas. Este fenómeno se explica en parte por la presencia de un ruido de fondo de medición que muestra ser tanto más preponderante en el cálculo del coeficiente de modulación cuanto más baja sea la expresión de los genes.
El análisis diferencial según el método llamado de Global Error Assessment (GEA), divulgado en el documento al que se ha hecho antes referencia, permite corregir este sesgo. Consiste en reagrupar los genes según un criterio estadístico, llamado significatividad (o p-value, en inglés), teniendo en cuenta la variabilidad del coeficiente de modulación en función del nivel de expresión en la condición de referencia para cada gen. La variabilidad del coeficiente de modulación representa, para un gen dado, la desviación típica de los coeficientes de modulación con el coeficiente de modulación medio. Este p-value traduce la significatividad de un valor de coeficiente de modulación. Esto permite obtener grupos de genes correspondientes a un p-value dado y equilibrar, en la lista de los genes seleccionados, la proporción de los genes débilmente expresados en la condición de referencia.
Sin embargo, el p-value no tiene sentido biológico. Por ello, el biólogo, que razona en el universo de las modulaciones, no puede apoyarse en este valor para identificar los genes diferenciados. Por consiguiente, no puede utilizar el método llamado de GEA para encontrar los genes diferenciados.
En la práctica, tras uno o más análisis diferenciales, el biólogo utiliza más frecuentemente técnicas de clasificación y de visualización con el fin de identificar genes que presentan perfiles de modulación de expresión similares entre varias condiciones. Se trata, por ejemplo, de la técnica de clasificación jerárquica o la clasificación por Descomposición Robusta en Valores Singulares divulgada en el documento L. Liu et al., Robust singular value decomposition analysis of microarray data, PNAS, 2003.
Sin embargo, en estas técnicas el biólogo se ve inducido, debido a las limitaciones ligadas a la visualización, o por concentrarse en análisis más complejos, tales como análisis de ontología o relativos a rutas metabólicas, a limitar el tamaño de las listas de genes seleccionados. También se apoya en los niveles de modulación de expresión medidos en cada condición, y no tiene, pues, en cuenta la significatividad asociada. Se pierde así la información relativa a esta significatividad durante la visualización de los niveles de modulación tras la clasificación. En otras palabras, el biólogo considera simplemente la razón del nivel de expresión de genes entre dos condiciones, clasificados por orden decreciente según su coeficiente de modulación. Se trata de la modulación estándar.
Deseando generalmente visualizar los genes más modulados en la condición de tratamiento, el biólogo aplica entonces una clasificación decreciente según el nivel de modulación y no conserva más que los primeros genes. Haciendo esto, no tiene en cuenta la significatividad y reintroduce el sesgo de selección que había sido eliminado por el cálculo del p-value.
Teniendo en cuenta lo que antecede, un problema que la invención se propone resolver es realizar un método corrector de tratamiento de resultados de experimentos transcriptómicos obtenidos por análisis diferencial, que tiene en cuenta la significatividad asociada a los valores de coeficiente de modulación, y cuyo resultado es además explotable a partir de valores que tienen un sentido biológico.
La solución propuesta por la invención a este problema tiene por objeto un método corrector de tratamiento de resultados de experimentos transcriptómicos obtenidos por análisis diferencial, consistente en las etapas siguientes:
caracterizado por incluir además una etapa de cálculo y de asociación de un coeficiente de modulación mediano sobre la curva isobárica de cada p-value observado.
Ventajosamente, dichas etapas de cálculo de un p-value asociado a cada coeficiente de modulación y de cálculo de un coeficiente de modulación mediano según la curva isobárica de cada p-value observado son realizadas por medio del método llamado GEA; dichas etapas de obtención de los resultados del nivel de expresión de genes en una condi- ción(es) de referencia, de obtención de los resultados del nivel de expresión de genes en una condición(es) de tratamiento, de cálculo del coeficiente de modulación (variación) del nivel de expresión, de cálculo del p-value y de cálculo de un coeficiente de modulación mediano sobre la curva isobárica de cada p-value observado son realizadas para una pluralidad de condiciones de tratamiento diferentes; dicha etapa de cálculo de curvas isobáricas de p-value en función del nivel medio de expresión de cada uno de dichos genes en la condición de referencia comprende una representación mediante un punto de cada gen estudiado sobre un gráfico que presenta, en la abscisa, el logaritmo del nivel medio de expresión en la condición de referencia indicado como x y, en la ordenada, el logaritmo del...
Reivindicaciones:
1. Método corrector llevado a cabo por ordenador de tratamiento de resultados de experimentos transcriptómicos obtenidos por análisis diferencial, consistente en las etapas siguientes:
- obtención de los resultados del nivel de expresión de genes en una condición de referencia y cálculo del nivel medio de expresión de cada uno de dichos genes;
- obtención de los resultados del nivel de expresión de dichos genes en una condición de tratamiento y cálculo del nivel medio de expresión para cada uno de dichos genes;
- cálculo del coeficiente de modulación del nivel de expresión para cada uno de dichos genes;
- cálculo de un p-value asociado a cada coeficiente de modulación;
- cálculo de curvas isobáricas de p-value en función del nivel medio de expresión de cada uno de dichos genes en la condición de referencia;
caracterizado por incluir además una etapa de cálculo y de asociación de un coeficiente de modulación mediano en la curva isobárica de cada p-value observado.
2. Método según la reivindicación 1, caracterizado por realizar dichas etapas de cálculo de un p-value asociado a cada coeficiente de modulación y de cálculo de un coeficiente de modulación mediano según la curva isobárica de cada p-value observado por medio del método llamado GEA.
3. Método según una de las reivindicaciones 1 ó 2, caracterizado por llevar a cabo dichas etapas de obtención de los resultados del nivel de expresión de genes en una condición de referencia, de obtención de los resultados del nivel de expresión de genes en una condición de tratamiento, de cálculo del coeficiente de modulación del nivel de expresión, de cálculo del p-value y de cálculo de un coeficiente de modulación mediano en la curva isobárica de cada p-value observado para una pluralidad de condiciones de tratamiento diferentes.
4. Método según una de las reivindicaciones 1, 2 ó 3, caracterizado por el hecho de que dicha etapa de cálculo de curvas isobáricas de p-value en función del nivel medio de expresión de cada uno de dichos genes en la condición de referencia comprende una representación mediante un punto de cada gen estudiado en un gráfico que presenta, en la abscisa, el logaritmo del nivel medio de expresión en la condición de referencia indicado como x y, en la ordenada, el logaritmo del nivel medio de expresión en la condición de tratamiento indicado como y, correspondiendo una curva isobárica de nivel p a los puntos teóricos para los cuales el p-value es igual a p.
5. Método según una de las reivindicaciones 1 a 4, caracterizado por el hecho de que un usuario selecciona genes de interés en base a un valor que tiene un sentido biológico.
6. Método según la reivindicación 5, caracterizado por el hecho de que dicho valor que tiene un sentido biológico es un valor de coeficiente de modulación.
7. Método según una de las reivindicaciones precedentes, caracterizado por el hecho de que un usuario selecciona genes de interés en base a un valor que tiene una significatividad.
8. Método según una de las reivindicaciones precedentes, caracterizado por realizar los experimentos transcriptómicos sobre chips de ADN.
9. Ordenador para la realización de un método corrector de tratamiento de resultados de experimentos transcriptómicos obtenidos por análisis diferencial según una de las reivindicaciones precedentes.
Patentes similares o relacionadas:
METODOS Y PRODUCTOS PARA GENOTIPADO IN VITRO, del 30 de Septiembre de 2010, de PROGENIKA BIOPHARMA, S.A.: El método de genotipado se basa en la combinación de un diseño experimental de DNA-chips de genotipado y en el desarrollo de un sistema secuencial de procesamiento e interpretación […]
 ANALISIS MATEMATICO PARA LA ESTIMACION DE CAMBIOS EN EL NIVEL DE EXPRESION GENICA, del 14 de Enero de 2010, de AVENTIS PHARMACEUTICALS INC.: Un método para calcular indicios de diferencias en el nivel de expresión génica en una pluralidad de hibridaciones de matrices, comprendiendo el método:
(a) determinar […]
ANALISIS MATEMATICO PARA LA ESTIMACION DE CAMBIOS EN EL NIVEL DE EXPRESION GENICA, del 14 de Enero de 2010, de AVENTIS PHARMACEUTICALS INC.: Un método para calcular indicios de diferencias en el nivel de expresión génica en una pluralidad de hibridaciones de matrices, comprendiendo el método:
(a) determinar […]
Sistemas y métodos para tratar, diagnosticar y predecir la aparición de una afección médica, del 15 de Julio de 2020, de Fundação D. Anna Sommer Champalimaud E Dr. Carlos Montez Champalimaud: Aparato para evaluar si es probable que un paciente tenga un estadio patológico favorable de cáncer de próstata, el aparato comprende: (a) un dispositivo de obtención […]
Dispositivo de procesamiento de datos para el procesamiento de valores de medición, del 22 de Abril de 2020, de F. HOFFMANN-LA ROCHE AG: Dispositivo de procesamiento de datos para el procesamiento de valores de medición de un equipo de medición médico, con una unidad de entrada , una […]
Sistema de análisis apuntador de acontecimientos y de gestión de medicación, del 30 de Octubre de 2019, de CAREFUSION 303, INC: Un método para reducir el riesgo de errores de medicación, el método comprende las etapas de: recibir en un dispositivo móvil un primer valor de un parámetro […]
Sistema y método de visualización en 2D y 3D para la inspección de un tubo de horno, del 11 de Septiembre de 2019, de Quest Integrity USA, LLC: Un sistema para la visualización de datos de inspección recogidos de un horno con una geometría física especificada, en donde dicho horno comprende […]
 Reconocimiento anatómico y análisis dimensional para asistir en la cirugía de mama, del 25 de Julio de 2019, de ALLERGAN, INC.: Método implementado por ordenador de determinación de mediciones de la mama de manera automática a partir de una representación tridimensional (3-D) de la parte superior […]
Reconocimiento anatómico y análisis dimensional para asistir en la cirugía de mama, del 25 de Julio de 2019, de ALLERGAN, INC.: Método implementado por ordenador de determinación de mediciones de la mama de manera automática a partir de una representación tridimensional (3-D) de la parte superior […]
Sistemas y métodos para tratar, diagnosticar y predecir la aparición de una afección médica, del 24 de Julio de 2019, de Fundação D. Anna Sommer Champalimaud E Dr. Carlos Montez Champalimaud: Un aparato para evaluar un riesgo de fallo clínico en un paciente después de que el paciente se haya sometido a una prostatectomía radical, y el aparato […]