ANALOGOS DE LAS AZINOMICINAS COMO AGENTES ANTI-TUMORES Y COMO PROMEDICAMENTOS.
Un compuesto de la formula I o una sal de la misma:
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/GB2006/000941.
Solicitante: UNIVERSITY OF BRADFORD.
Nacionalidad solicitante: Reino Unido.
Dirección: RICHMOND ROAD,BRADFORD BD7 1DP.
Inventor/es: SCARCEY,MARK, PATTERSON,LAURENCE HYLTON INST. OF CANCER THERAP, PORS,KLAUS INST. OF CANCER THERAP, CASELY-HAYFORD MAXWELL.
Fecha de Publicación: .
Fecha Concesión Europea: 18 de Noviembre de 2009.
Clasificación Internacional de Patentes:
- C07C235/28 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07C COMPUESTOS ACICLICOS O CARBOCICLICOS (compuestos macromoleculares C08; producción de compuestos orgánicos por electrolisiso electroforesis C25B 3/00, C25B 7/00). › C07C 235/00 Amidas de ácidos carboxílicos, estando sustituida la estructura carbonada de la parte ácida por átomos de oxígeno. › siendo la estructura carbonada acíclica e insaturada.
- C07C69/94 C07C […] › C07C 69/00 Esteres de ácidos carboxílicos; Esteres del ácido carbónico o del ácido halofórmico. › de ácidos hidroxicarboxílicos policíclicos cuyos grupos hidroxilo y carboxilo están unidos a átomos de carbono de ciclos aromáticos de seis miembros.
- C07D211/38 C07 […] › C07D COMPUESTOS HETEROCICLICOS (Compuestos macromoleculares C08). › C07D 211/00 Compuestos heterocíclicos que contienen ciclos hidrogenados de piridina, no condensados con otros ciclos. › Atomos de halógeno o radicales nitro.
- C07D211/42 C07D 211/00 […] › unidos en posición 3 ó 5.
- C07D303/46 C07D […] › C07D 303/00 Compuestos que contienen ciclos de tres miembros que tienen un átomo de oxígeno como único heteroátomo del ciclo. › por radicales amida o nitrilo.
- C07D405/12 C07D […] › C07D 405/00 Compuestos heterocíclicos que contienen a la vez uno o más heterociclos que tienen átomos de oxígeno como únicos heteroátomos del ciclo y uno o más heterociclos que tienen átomos de nitrógeno como único heteroátomo del ciclo. › unidos por una cadena que contiene heteroátomos como enlaces de cadena.
Clasificación PCT:
- A61K31/4523 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › conteniendo otros sistemas heterocíclicos.
- A61P35/00 A61 […] › A61P ACTIVIDAD TERAPEUTICA ESPECIFICA DE COMPUESTOS QUIMICOS O DE PREPARACIONES MEDICINALES. › Agentes antineoplásicos.
- C07C69/94 C07C 69/00 […] › de ácidos hidroxicarboxílicos policíclicos cuyos grupos hidroxilo y carboxilo están unidos a átomos de carbono de ciclos aromáticos de seis miembros.
- C07D211/38 C07D 211/00 […] › Atomos de halógeno o radicales nitro.
- C07D211/42 C07D 211/00 […] › unidos en posición 3 ó 5.
- C07D303/46 C07D 303/00 […] › por radicales amida o nitrilo.
- C07D405/12 C07D 405/00 […] › unidos por una cadena que contiene heteroátomos como enlaces de cadena.
Fragmento de la descripción:
Análogos de las azinomicinas como agentes anti-tumores y como promedicamentos.
Campo de la invención
La presente invención se refiere a promedicamentos activados por oxidación aromática, especialmente promedicamentos anti-tumores y aquellos activados por las actividades de oxidación de la familia de enzimas citocromo P450. Los promedicamentos pueden ser agentes alcilativos con actividades de inhibición de topoisómeros II.
Se conocen muchas drogas citotóxicas convencionales que pueden ser usadas con fines terapéuticos. Sin embargo, generalmente sufren del problema que son, por lo general, citotóxicas y, en consecuencia, pueden afectar a células distintas a aquéllas cuya destrucción se requiere. Esto puede ser aliviado hasta cierta medida por el uso de sistemas de distribución dirigida de drogas, por ejemplo, inyección a una zona de tejido tumoroso, o, por ejemplo, enlazando el agente citotóxico a un anticuerpo que reconoce específicamente un antígeno que se despliegue únicamente sobre la superficie celular cancerosa. Alternativamente, puede usarse radiación electromagnética para provocar alteración química en una agente en una zona deseada de tal forma que se convierta en citotóxica. Sin embargo, todas estas técnicas tienen, en mayor o menor medida, ciertas limitaciones y desventajas.
Las azinomicinas A y B son potentes agentes anti-tumores que se enlazan al ADN por acilación en el canal principal y llevan a la muerte celular. Sin embargo, son relativamente inestables, son escasamente disponibles de fuentes naturales y es improbable su desarrollo en el ámbito clínico.
Antecedentes de la invención
Estos compuestos que se forman naturalmente, además del análogo A truncado (vid. estructura infra), fueron aislados del estreptomices griseofuscus S42227 por Nagaoka y otros en Japón (J. Antibiot. (Tokio) 1986, 39, 1527-1532).
Armstrong en Tetrahedron Lett. 1991, 32, 3807-3810, descubrió posteriormente, usando datos especiales NMR y masivos, que el antibiótico anti-tumor carzinofilina, aislado en 1954 del estreptomices sahaquiror (Onda y otros, J. Antibiot. 1969, 22, 42-44) era de la misma composición que el producto natural azinomicina B.
Shibuya en Tetrahedron Lett. 1983, 24, 1175-1178, describe los primeros estudios sintéticos de las azinomicinas, pero estos son inexactos, en cuanto se basan en la estructura errónea de carzinofilina sugerida por Lain y otros en J. Am. Chem. Soc. 1982, 104, 3213-3214.
El análogo truncado A fue sintetizado correctamente por primera vez por Shibuya y otros en Tetrahedron Lett. 1987, 28, 2619-2622, donde la diacetona D-glucosa de la reserva quiral, disponible comercialmente, se usaba en una síntesis larga, de múltiples etapas, para generar estereoespecíficamente el análogo A, de la estructura que se muestra supra, con la misma estereoquímica que los productos naturales.
La mayoría de los otros estudios sobre fragmento epóxido de las azinomicinas y sobre la síntesis de A se han enfocado en el uso de epoxidación asimétrica sin ángulos agudos. Los esfuerzos directos sobre precursores enantiopuros sintetizadores son descritos por Konda y otros en Chem. Pharmac. Bull 1994, 42, 285-288. Shipman y otros en Chem. Soc. Perkin Trans. 11998, 1249-1255, discuten ulteriormente una metodología de dihidroxilación asimétrica/epoxidación asimétrica para obtener el isómero S,S requerido con una producción excelente.
Tanto Amstrong y otros (J. Am. Chem. Soc. 1992, 114, 371-372) y Coleman y otros (J. Org. Chem. 1992, 57, 5813-5815) han descrito independientemente rutas sintéticas al núcleo aziridino de la Azinomicina A. La síntesis total se mostró más elusiva y sólo ha sido recientemente descrita por Coleman y otros (Angew. Chem. Int. Ed. 2001, 491736-1739). La clave hacia la síntesis total fue un ensamblaje de la parte esencial del producto natural, incluyendo la parte epóxida, seguido de la tardía introducción de etapas del sistema azabicíclico por una reacción tipo Wadsworth-Homer-Emmons.
La síntesis del fragmento de la izquierda de las azinomicinas permitió el estudio de sus interacciones con el ADN. Zang y otros en Biochemistry 2000, 39, 14968-14975 presentan datos que sugieren que la estructura A se intercala con el ADN por la vía de su subunidad de naftalina y de sus residuos de guanina de alcilatos en N7, con poca selectividad de secuencias, si es que hay alguna. Shipman y otros usaron estos descubrimientos en investigaciones de actividad de estructuras para identificar análogos de los productos naturales que podrían ser útiles como agentes anti-tumores (Bioorg. Med. Chem. Lett 2000, 10, 239-241). La sustitución del 3-metóxido-5-metilnaftalina con un grupo fenilo (del que se podría esperar que muestre poca afinidad con el ADN por intercalación) eliminaría de forma efectiva la potencia biológica del epóxido en una variedad de líneas celulares. En Chem. Commun. 2000, 2325-2326, Hortley y otros estudian actividad de entrelazamiento de ADN de dimeros simétricos basada en el dominio de epóxido de las azinomicinas. Demostraron que que una longitud de enlazamiento óptima parecía ser la de 4 grupos de metileno y que los agentes pueden entrelazarse con el ADN, y tener una potente actividad citotóxica, aunque ninguno de los componentes tuvieron una actividad significativamente superior que el A que se entrelaza.
Las azinomicinas parecen actuar interrumpiendo la replicación celular del ADN por formación de entrelazamiento filamentoso. Lain y otros en Can. J. Biochem. 1997, 55, 630-635, fueron los primeros en notar la aptitud de la azinomicina B para formar lazos covalentes entre filamentos complementarios de ADN. Fujiwara y otros en Tetrahedron Lett. 1999, 40, 315-318 sugieren, además, que el entrelazamiento tiene lugar por la vía de una alcilación inicial de la aziridina con el N7 de adenina seguida por el entrelazamiento eficiente a través de una segunda reacción del N7 de una guanina 2 que se basa lejanamente en el epóxido.
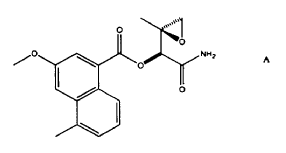
Casely-Hayford y otros en Bioorganic and Med. Chem. Letters (2005) 15, 653-656, discuten el diseño y la síntesis de un análogo B de azinomicina, potencialmente viable en terapia, basado en A implicando el emparejamiento de una mostaza de piperidina con el clorido ácido del cromoforo de azinomicina.
Los autores concluyen con que la monoalcilación es suficiente para la actividad biológica y que el entrelazado puede incluso ser perjudicial.
La presente invención se refiere al uso terapéutico primario de una gama de análogos de la azinomicina y su síntesis. Los compuestos que se incorporan aquí son nuevos. La presente invención también se relaciona con precursores sintéticos de análogos de azinomicina que no tienen el anillo de epóxido o de la aziridina de los productos naturales, y que por sí mismos son sustancialmente inactivos como agentes alcilantes del ADN.
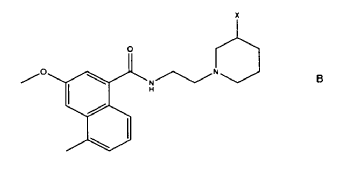
Se ha informado (Murray y otros, 1997, Cancer Research, 57, 3026-3031 y Patente Mundial A-9712246) que la enzima CYP1B1, miembro de la familia cicotrónica P450 (CYP) de enzimas metabolizantes xenobióticos, aparece de forma muy frecuente en una variedad de cánceres humanos, incluyendo cánceres de mama, colon, pulmón, esófago, piel, nudos linfáticos, cerebro y testículos, y que no se detecta en tejido normal. Esto llevó a la conclusión que la expresión de isoformas P450 de citocromo en células tumorosas proporciona un objetivo molecular para el desarrollo de nuevos medicamentos anti-tumores que podrían ser activadas selectivamente por las encimas CYP en células tumorosas, aunque no se dan ejemplos de medicamentos. Muchos de los CYPs expresados en tumores se mencionan en Patterson, LH y otros (1999) Anticancer Drug Des. 14 (6) 473-486.
En la Patente Mundial 02/067930A1, Searcey y Petterson describen diversos componentes de benz-indola y benzo-quinolina como pro-medicamentos para tratamiento de tumores. En la Patente 02/068412A1 describen también derivados de pirrolo-índola y pirrolo-quinolina para su uso con pro-medicamentos oxidables-CYP y en la Patente Mundial 02/067937A1 se describen pro-medicamentos oxidables-CYP de indolita y tetrahidro-quinolina. De todos estos compuestos se espera su hidroxilatación en el átomo de carbono al que X está unido por...
Reivindicaciones:
1. Un compuesto de la formula I o una sal de la misma:
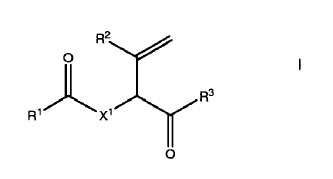
En el que X1 se selecciona de un grupo consistente en O, S y NR0 en el que R0 es alcilo C1-4 ó H;
R3 es NH2, NHR5, SR4, OR4, CH2R4 u OH;
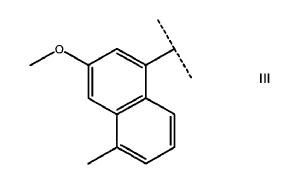
R1 R6 es naftilo, antranilo o un grupo de la formula III, opcionalmente sustituidos
R2 es H, C1-4-alcílico opcionalmente sustituido, C1-4-alcóxido, fenilo opcionalmente sustituido, C7-12-arálcilo, heterorarilo o un enlazante opcionalmente sustituidos;
R4 es C1-4-alcílico, C1-4-alcílico sustituido, C1-4-alcóxido, fenilo opcionalmente sustituido, C7-12-arálcilo, heterorarilo, CnH2nNR5R6 o un enlazante opcionalmente sustituidos;
En el que al menos uno de R5 y R1 es (CH2)2A2 o conjuntamente con el nitrógeno al que está agregado forma un anillo de formula II
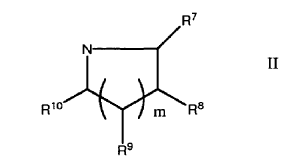
En el que al menos uno de R7, R8 y R9 se selecciona de A1 y A1, C1-4-alcílico sustituido, y algunos otros son H ó C1-4-alcílico; R10 se selecciona de H, C1-4-alcílico, A1 y A1, C1-4-alcílico sustituido;
A1 es un grupo de salida o un átomo de halógeno;
m es 1-4;
n es 1-7;
siendo seleccionados los grupos sustituyentes de C1-4-alcílico, hidróxilo, amino, alcilamino, halo, y aziridina, y el enlazante es un oligopéptido, biotina, avidina, estreptavidina o grupo polimérico, un oligonucleótido o una proteína.
2. Un compuesto de acuerdo con la reivindicación 1 en la que X1 es O.
3. Un compuesto de acuerdo con la reivindicación 1 ó la reivindicación 2 en la que R2 es CH3.
4. Un compuesto de acuerdo con la reivindicación 1-3 en la que R3 es NHR4.
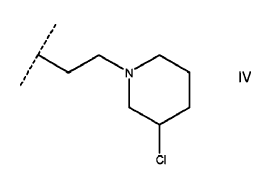
5. Un compuesto de acuerdo con la reivindicación 4 en el que R4 es CnH2nNR5R6.
6. Un compuesto de acuerdo con la reivindicación 5 en el que R4 es CnH2nNR5R6 es IV
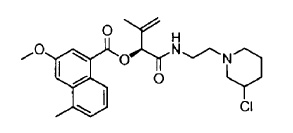
7. Un compuesto de acuerdo con la reivindicación 1 que es
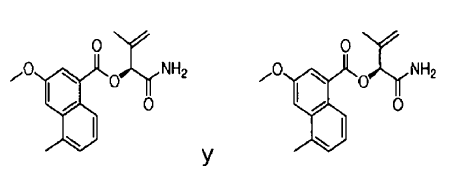
8. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1-3 en el que R3 es NH2.
9. Un compuesto de acuerdo con la reivindicación 8 seleccionado de
10. Un compuesto de acuerdo con cualquiera de las reivindicaciones anteriores para su uso en un método de tratamiento médico terapéutico de un animal.
11. El uso de un compuesto de acuerdo con cualquiera de las reivindicaciones 1-9 en la fabricación de una composición para uso en un método de tratamiento médico terapéutico de un animal, preferentemente un tratamiento antitumoroso.
12. Una composición farmacéutica que comprenda el compuesto de cualquiera de las reivindicaciones 1-9 y un excipiente farmacéuticamente aceptable.
13. Un método sintético en que un compuesto de fórmula V
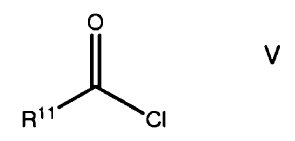
en el que R11 se selecciona de un grupo consistente de neftilo, antranilo y un grupo de fórmula III, opcionalmente sustituidos, se reactiva con un compuesto de fórmula VI
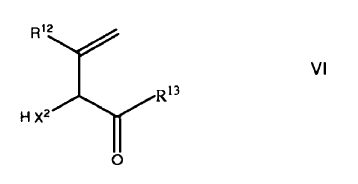
en el que R12 es H, C1-4-alcílico, opcionalmente sustituido, C1-4-alcóxido, fenilo, opcionalmente sustituido, C7-12-arálcilo, heterorarilo o un enlazante, opcionalmente sustituidos;
X2 es O, NH o S;
R13 es OH, Cl, C1-4-alcóxido u OPG donde PG es un grupo protector;
Tal que Cl en V es reemplazado en una reacción de sustitución nucleofílica por un grupo de fórmula VII

donde el enlazante es un oligopéptido, biotina, avidita, estreptavidina, un grupo polimérico, un oligonucleótido o una proteína.
14. Un método de acuerdo con la reivindicación 13 seguido por eliminación del grupo protector para conseguir R13 = OH.
15. Un método de acuerdo con la reivindicación 14 seguido por reacción con un grupo de la fórmula HR14 para dar un compuesto de formula VIII
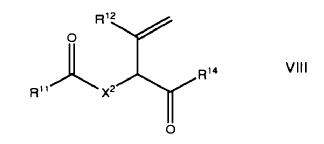
en el que R14 se selecciona del grupo consistente en NH2, NH315, SR15 y OR15;
R15 es C1-4-alcílico, C1-4-alcílico sustituido, C1-4-alcóxido, fenilo opcionalmente sustituido, C7-12-arálcilo, heterorarilo, CpH2pNR16R17 o un enlazante, opcionalmente sustituidos;
En el que al menos uno de R16 y R17 es (CH2)2A2 o conjuntamente con el nitrógeno al que está agregado forma un anillo de formula IX
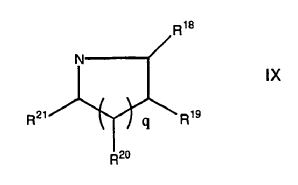
En el que al menos uno de R18, R19 y R20 se selecciona de A2 y A2 C1-4-alcílico sustituido, y algunos otros son H ó C1-4-alcílico;
R21 se selecciona de H, C1-4-alcílico, A y A C1-4-alcílico sustituido;
A2 es un grupo de salida, átomo de halógeno, hidroxilo o un hidroxilo protegido;
q es 1-4;
p es 1-7;
siendo los grupos sustituidos seleccionados de C1-4-alcílico, hidróxilo, amino, alcilamino, halo, y aziridina.
16. Un método de acuerdo con cualquiera de las reivindicaciones 13-15 donde el grupo protector en bencilo.
17. Un método de acuerdo con cualquiera de las reivindicaciones 13-16 en el que X2 es O.
18. Un método de acuerdo con cualquiera de las reivindicaciones 13-17 en el que R12 es CH3.
19. Un método de acuerdo con cualquiera de las reivindicaciones 13-18 en el que el alcino al que R12 está agregado es oxidado para conseguir el epóxido correspondiente.
20. Un compuesto de la fórmula general X
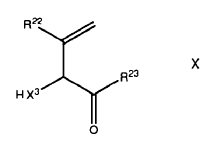
en el que X3 se selecciona del grupo consistente en O, NH y S;
R22 es H, C1-4-alcílico, C1-4-alcóxido, fenilo opcionalmente sustituido, C7-12-arálcilo, heterorarilo o un enlazante, opcionalmente sustituidos;
R23 es un enlazante o NHR24 donde R24 es CrH2rNR25R26 o un enlazante;
R25 y R26 son (CH2)2A3 o conjuntamente con el nitrógeno al que está agregado forma un anillo de formula XI
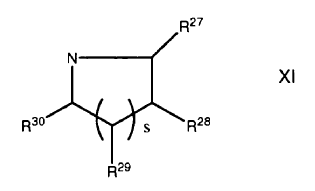
En el que al menos uno de R27, R28 y R29 se selecciona se selecciona de A3 y A3, C1-4-alcílico sustituido, y algunos otros son H ó C1-4-alcílico; R30 se selecciona de H, C1-4-alcílico, A3 y A3 C1-4-alcílico sustituido;
A3 es un grupo de salida, átomo de halógeno, hidroxilo o hidroxilo protegido;
s es 1-4;
r es 1-7 y el enlazante es un oligopéptido, biotina, avidita, estreptavidina, un grupo polimérico, un oligonucleótido o una proteína.
Patentes similares o relacionadas:
Formas cristalinas de 6-((6,7-dimetoxiquinazolin-4-il)oxi)-N,2-dimetilbenzofuran-3-carboxamida, del 29 de Julio de 2020, de Hutchison Medipharma Limited: Forma I de 6-((6,7-dimetoxiquinazolin-4-il)oxi)-N,2-dimetilbenzofuran-3-carboxamida, en donde el difractograma de rayos X de polvo de la Forma […]
Compuestos heterocíclicos que activan AMPK y métodos de uso de los mismos, del 29 de Julio de 2020, de RIGEL PHARMACEUTICALS, INC.: Un compuesto que es N-((cis)-1-(4-cianobencil)-3-fluoropiperidin-4-il)-6-(4-(4-metoxibenzoil)piperidin-1-carbonil)nicotinamida; N-((3S,4S)-1-(4-cianobencil)-3-fluoropiperidin-4-il)-6-(4-(4-metoxibenzoil)piperidin-1-carbonil)nicotinamida; […]
Derivados de piperidina 1,4 sustituidos, del 29 de Julio de 2020, de 89Bio Ltd: Un compuesto de acuerdo con la Fórmula I: **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en donde: A se selecciona de […]
Ureas asimétricas p-sustituidas y usos médicos de las mismas, del 22 de Julio de 2020, de Helsinn Healthcare SA: Un compuesto de Fórmula I: **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en donde: una línea discontinua indica un enlace opcional; X es CH; […]
Compuestos utilizados como inhibidores de la quinasa reordenada durante la transfección (RET), del 1 de Julio de 2020, de GlaxoSmithKline Intellectual Property Development Limited: Un compuesto de acuerdo con la Fórmula (I), o una sal farmacéuticamente aceptable del mismo: **(Ver fórmula)** en donde: X es N o CR5; Y es un enlace; […]
Forma cristalina de un derivado de benzimidazol y un método de preparación del mismo, del 24 de Junio de 2020, de HK INNO.N CORPORATION: Una forma cristalina A de un compuesto representado por la siguiente Fórmula 1 que tiene un patrón de difracción de rayos X en polvo usando radiación Cu-Kα, con picos […]
Compuestos de diaminopirimidilo sustituidos, composiciones de los mismos y procedimientos de tratamiento con ellos, del 17 de Junio de 2020, de SIGNAL PHARMACEUTICALS LLC: Un compuesto de fórmula (I): **(Ver fórmula)** o una sal, un tautómero, un isotopólogo o un estereoisómero farmacéuticamente aceptable […]
Derivados de N-piperidinil acetamida como bloqueadores de canales de calcio, del 20 de Mayo de 2020, de Praxis Precision Medicines, Inc: Una composición farmacéutica que comprende un compuesto que tiene la estructura: **(Ver fórmula)** o una sal o conjugado farmacéuticamente […]