Uso de aptámeros en proteómica.
Un método para medir la cantidad de al menos una molécula en una muestra biológica,
comprendiendo el método:
a) combinar la muestra con uno o más aptámeros y permitir que una o más moléculas en la muestra se unan al o a los aptámeros;
b) separar las moléculas unidas de las no unidas; y
c) cuantificar la o las moléculas unidas al o a cada uno de los aptámeros,
en donde la cuantificación de la o las moléculas unidas se lleva a cabo secuenciando al menos parte del o de cada uno de los aptámeros.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/GB2008/003447.
Solicitante: PRONOTA NV.
Nacionalidad solicitante: Bélgica.
Dirección: VIB BIO-INCUBATOR TECHNOLOGIEPARK 4 9052 ZWIJNAARDE GHENT BELGICA.
Inventor/es: BROWN,CLIVE GAVIN, KAS,KOEN, EYCKERMAN,SVEN AGNES JAN.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C12N15/11 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12N MICROORGANISMOS O ENZIMAS; COMPOSICIONES QUE LOS CONTIENEN; PROPAGACION, CULTIVO O CONSERVACION DE MICROORGANISMOS; TECNICAS DE MUTACION O DE INGENIERIA GENETICA; MEDIOS DE CULTIVO (medios para ensayos microbiológicos C12Q 1/00). › C12N 15/00 Técnicas de mutación o de ingeniería genética; ADN o ARN relacionado con la ingeniería genética, vectores, p. ej. plásmidos, o su aislamiento, su preparación o su purificación; Utilización de huéspedes para ello (mutantes o microorganismos modificados por ingeniería genética C12N 1/00, C12N 5/00, C12N 7/00; nuevas plantas en sí A01H; reproducción de plantas por técnicas de cultivo de tejidos A01H 4/00; nuevas razas animales en sí A01K 67/00; utilización de preparaciones medicinales que contienen material genético que es introducido en células del cuerpo humano para tratar enfermedades genéticas, terapia génica A61K 48/00; péptidos en general C07K). › Fragmentos de ADN o de ARN; sus formas modificadas (ADN o ARN no empleado en tecnología de recombinación C07H 21/00).
- C12N15/115 C12N 15/00 […] › Aptámeros, p.ej. ácidos nucleicos que unen una molécula diana específicamente y con alta afinidad sin hibridar entre ellos.
- G01N33/68 FISICA. › G01 METROLOGIA; ENSAYOS. › G01N INVESTIGACION O ANALISIS DE MATERIALES POR DETERMINACION DE SUS PROPIEDADES QUIMICAS O FISICAS (procedimientos de medida, de investigación o de análisis diferentes de los ensayos inmunológicos, en los que intervienen enzimas o microorganismos C12M, C12Q). › G01N 33/00 Investigación o análisis de materiales por métodos específicos no cubiertos por los grupos G01N 1/00 - G01N 31/00. › en los que intervienen proteínas, péptidos o aminoácidos.
PDF original: ES-2448426_T3.pdf
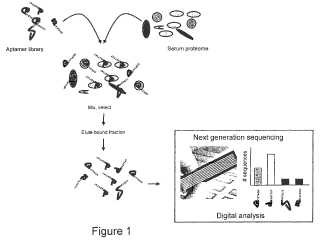
Fragmento de la descripción:
Uso de aptámeros en proteómica La presente invención se refiere a la identificación y cuantificación de proteínas en una mezcla compleja. En particular, la invención se refiere al uso de aptámeros como etiquetas o representantes para proteínas y cantidades de proteínas en una mezcla compleja. En otro aspecto, la invención se refiere al uso de métodos de secuenciación de polinucleótidos de la siguiente generación en proteómica.
El proteoma se describe habitualmente como el complemento completo de proteínas que se encuentra en un sistema biológico tal como, p. ej., una célula, tejido, fluido corporal, órgano u organismo. El estudio de proteínas que se producen de forma natural se denomina generalmente ‘proteómica’ y abarca el estudio del proteoma expresado en instantes particulares y/o bajo condiciones internas o externas de interés. Las estrategias de la proteómica tienen generalmente como objetivo el análisis global del proteoma y requieren que grandes números de proteínas,
p. ej., cientos o miles, puedan ser resueltos, identificados y cuantificados de forma rutinaria a partir de una sola muestra o de múltiples muestras.
Entre las promesas de la proteómica se encuentra su capacidad de reconocer nuevos biomarcadores, es decir, proteínas en calidad de indicadores biológicos que señalizan un estado fisiológico alterado, por ejemplo debido a una enfermedad o intervención terapéutica. El descubrimiento de biomarcadores implica habitualmente comparar proteomas expresados en distintos estados fisiológicos e identificar proteínas cuyos niveles de aparición o expresión difieran de forma consistente entre los estados fisiológicos (Schrattenholz A, Groebe K. Electrophoresis. (2007) Junio 28 (12) 1970-9) .
Las proteínas en la sangre son una diana particular para la identificación de marcadores de estados patológicos y tratamientos farmacológicos. Se asumen ampliamente que las cantidades y/o la conformación de proteínas en la sangre debería estar estadísticamente relacionada con estos estados de una manera que sopese la variabilidad natural intrínseca. La sangre y otros fluidos corporales son una diana particular, ya que bañan a los tejidos afectados, transportan proteínas vitales y pueden obtenerse para el ensayo utilizando procesos relativamente económicos y directos durante una consulta médica.
Sin embargo, las proteínas en la sangre tienen un intervalo muy amplio de concentraciones, contabilizando un número pequeño de proteínas más del 99, 9% de todas las proteínas y ocupando el resto una distribución desde pìcogramo a miligramo por mililitro (Qian W.J. et al., Mol. Cell. Prot. (2006) 5 (10) 1727-1744) . Debido a las limitaciones de las técnicas proteómicas existentes, este intervalo de abundancia sigue siendo una hipótesis. Los científicos de la proteómica han empleado una diversidad de métodos para alcanzar este intervalo, con el objetivo de minimizar también la alteración de las abundancias relativas de las proteínas. A menudo, esto requiere la exclusión de la elevada abundancia de proteínas mediante purificación selectiva. También se han realizado intentos para reducir la complejidad de los péptidos obtenidos, centrándose en subconjuntos de todos los péptidos en la muestra. Estos procesos son largos y todavía ha de demostrarse la reproducibilidad entre muestras, réplicas, máquinas y laboratorios de una manera que sea un requisito previo para el descubrimiento estadístico de proteínas biomarcadoras.
La proteómica basada en la espectrometría de masas (MS) convencional, habitualmente utilizada en el descubrimiento de biomarcadores, procede separando muestra biológicas para aislar proteínas individuales de la mezcla sometida a investigación. Más recientemente, esto ha avanzado de geles en 2D a cromatografía líquida de alta resolución (HPLC) basada en columna multidimensional. Las proteínas pueden disgregarse en subunidades más cortas o péptidos. Los péptidos aislados son luego alimentados a un espectrómetro de masas, el cual ioniza los péptidos y los disgrega adicionalmente, proporcionando una escalera de mediciones de masa/carga. Estas mediciones y sus abundancias también se pueden cuantificar bajo una diversidad de esquemas, habitualmente relacionados con un cierto control. La escalera de espectros resultante es interpretada después frente a secuencias de péptidos conocidas o en ciego a partir de datos en bruto, y la información sobre la masa y las secuencias obtenidas se utiliza para la búsqueda de bases de datos de secuencias para identificar las proteínas a partir de las cuales se originaron los péptidos respectivos.
Sin embargo, la proteolisis de muestras biológicas complejas produce habitualmente cientos de miles de péptidos que pueden superar la capacidad de resolución de sistemas cromatográficos y de MS conocidos, provocando una resolución incompleta y una identificación perjudicada de los péptidos constituyentes. Típicamente, en proteómicas basadas en MS, tanto como un 80% de los espectros derivados de la muestra no pueden ser re-interpretados de forma fiable o consistente en péptidos discriminatorios o, por lo tanto, proteínas. Su comportamiento en la fragmentación y abundancia en el proceso MS también puede depender del contexto, complicando adicionalmente la reproducibilidad (Liu H, Sadygov RG, Yates JR 3º, Anal. Chem. (2004) Jul 15 76 (14) 4193-201) .
Un método para permitir el análisis proteómico de muestras biológicas consiste en reducir la complejidad de mezclas de péptidos generadas por la separación de este tipo de muestras, antes de someter dichas mezclas de péptidos a etapas de resolución e identificación aguas abajo tales como separación cromatográfica y/o espectrometría de masas (MS) . De manera ideal, la reducción de la complejidad de las mezclas de proteínas y péptidos disminuirá el número medio de péptidos distintos presentes por proteína individual de la muestra, pero maximizará la fracción de proteínas de la muestra realmente representada en la mezcla de péptidos.
El uso de sangre (suero o plasma) se ve oscurecido adicionalmente por el procesamiento biológico de proteínas de una diversidad de modos que confunden la verificación basada en MS (Qian W.J. et al., Mol. Cell. Prot. (2006) 5 (10) 1727-1744) . Estudios recientes han mostrado intentos incontroladamente contradictorios de identificar y recontar proteínas a partir de muestras clínicas. El propio acto de aislamiento, fragmentación y medición de proteínas en la proteómica basada en MS convencional altera su abundancia relativa y constitución química.
El resultado del análisis basado en proteómica es una “lista de éxitos” de proteínas que están significativamente correlacionadas en las muestras sometidas a ensayo. Típicamente, la lista es una selección de proteínas de importancia estadística variable. Habitualmente se realiza un estudio biológicamente orientado de la lista y se hace una elección racional sobre la importancia biológica potencial de cada uno de los miembros. El proceso de alcanzar una lista de biomarcadores hipotéticos o de proteínas de importancia supuestamente estadística se denomina generalmente “Descubrimiento”.
Los esfuerzos están entonces enfocados hacia la verificación y validación de un pequeño número de “éxitos” elegidos. Esto implica la confirmación de las abundancias de proteínas medidas en una población más amplia de muestras clínicas, siendo el objetivo demostrar que las proteínas descubiertas y elegidas son genuinas y no falsas positivas y que son específicas para la enfermedad o estado médico. A este proceso de confirmar la importancia cuantitativa de una medición proteómica y de un supuesto biomarcador en una población más generalizada se le alude habitualmente como ‘Validación’ (Zolg, Mol. Cell. Prot. (2006) 5 (10) , 1720-1726) . A menudo esto requiere el uso de una tecnología alternativa a la fase de descubrimiento.
Típicamente, esto se consigue utilizando anticuerpos dirigidos contra las supuestas proteínas purificadas. Estos anticuerpos pueden luego utilizarse en un ensayo basado en ELISA contra cientos de muestras, re-aplicarse la estadística relevante y establecerse la validez de la proteína en calidad de biomarcador. La intención final de este proceso es también que el anticuerpo se vuelva parte de un análisis clínico. Sin embargo, la producción de los anticuerpos lleva mucho tiempo y es costosa, y no siempre es posible dirigir un anticuerpo específico a una proteína dada. Una complicación adicional es la presencia de variantes alternativas de la proteína o isoformas que puedan tener incrustaciones de moléculas de azúcar fijadas y otras modificaciones... [Seguir leyendo]
Reivindicaciones:
1. Un método para medir la cantidad de al menos una molécula en una muestra biológica, comprendiendo el
método: a) combinar la muestra con uno o más aptámeros y permitir que una o más moléculas en la muestra se unan al o a los aptámeros;
b) separar las moléculas unidas de las no unidas; y
c) cuantificar la o las moléculas unidas al o a cada uno de los aptámeros,
en donde la cuantificación de la o las moléculas unidas se lleva a cabo secuenciando al menos parte del o de cada uno de los aptámeros.
2. Un método de acuerdo con la reivindicación 1, en el que la al menos una molécula es una proteína.
3. Un método de acuerdo con la reivindicación 1 o la reivindicación 2, en el que se conoce la identidad de la molécula.
4. Un método de acuerdo con la reivindicación 1 o la reivindicación 2, en el que se desconoce la identidad de la molécula.
5. Un método de acuerdo con la reivindicación 4, en el que dicho método comprende, además, determinar la identidad de la molécula.
6. Un método de acuerdo con una cualquiera de las reivindicaciones 1 a 5, en el que se conoce la secuencia del o de cada uno de los aptámeros.
7. Un método de acuerdo con una cualquiera de las reivindicaciones 1 a 6, en el que cada una de las secuencias de los aptámeros porta una etiqueta única.
8. Un método de acuerdo con la reivindicación 7, en el que la etiqueta es la secuencia del aptámero.
9. Un método de acuerdo con la reivindicación 7 o la reivindicación 8, en el que la etiqueta es parte de la secuencia del aptámero.
10. Un método de acuerdo con una cualquiera de las reivindicaciones 1 a 9, en el que la secuenciación se lleva a cabo en una disposición ordenada de una molécula sencilla o en una disposición ordenada de una molécula sencilla clonal.
11. Un método de acuerdo con una cualquiera de las reivindicaciones 1 a 10, en el que el método comprende, además, separar el o los aptámeros unidos a la o a cada una de las moléculas y disponer ordenadamente el o los aptámeros sobre una superficie.
12. Un método de acuerdo con la reivindicación 11, en el que el método comprende, además, amplificar los aptámeros dispuestos ordenadamente.
13. Un método de acuerdo con una cualquiera de las reivindicaciones 1 a 12, en el que el uno o más aptámeros comprenden diferentes secuencias que se unen a la misma molécula.
14. Un método de acuerdo con una cualquiera de las reivindicaciones 1 a 12, en el que el uno o más aptámeros comprenden paneles de secuencias de aptámeros, uniéndose cada uno de los paneles a una molécula diferente.
15. Un método de acuerdo con una cualquiera de las reivindicaciones 1 a 14, en el que los aptámeros se derivan de polinucleótidos.
16. Un método de acuerdo con la reivindicación 15, en el que los aptámeros son polinucleótidos y tienen entre aproximadamente 30 y aproximadamente 60 bases.
17. Un método de acuerdo con la reivindicación 16, en el que los aptámeros tienen entre aproximadamente 40 bases.
18. Un método de acuerdo con una cualquiera de las reivindicaciones 1 a 17, en el que la muestra biológica es un fluido corporal.
19. Un método de acuerdo con la reivindicación 18, en el que el fluido corporal es sangre o se deriva de sangre.
20. Un método de acuerdo con la reivindicación 19, en el que el fluido corporal es suero o plasma.
21. Un método de acuerdo con una cualquiera de las reivindicaciones 1 a 20, en el que el método comprende, además: 5 d) combinar una segunda muestra biológica con la fracción de aptámero unida, obtenida en c) ; e) separar moléculas unidas de las no unidas; f) cuantificar la o las moléculas unidas a aptámeros; y g) comparar las cantidades obtenidas en c) con las obtenidas en f) ,
en donde la cuantificación de aptámeros unidos a una o más moléculas en la segunda muestra se lleva a cabo 10 secuenciando al menos parte de cada uno de los aptámeros.
22. Un método de acuerdo con una cualquiera de las reivindicaciones 1 a 21, en el que el método comprende, además comparar la cantidad del o de cada uno de los aptámeros frente a una cantidad control o de línea base.
23. Un método de acuerdo con la reivindicación 21 o la reivindicación 22, en el que la segunda muestra se obtiene de un individuo del que se sabe que se encuentra en un estado patológico.
24. Un método de acuerdo con la reivindicación 21 o la reivindicación 22, en el que la segunda muestra se obtiene de un individuo del que se sabe que se encuentra en un estado sano.
25. Un método de acuerdo con la reivindicación 21 o la reivindicación 22, en el que la segunda muestra se obtiene de un individuo después de tratamiento con fármacos.
26. Uso de un método según una cualquiera de las reivindicaciones 1 a 25, para identificar uno o más 20 biomarcadores.
27. Uso de un método según una cualquiera de las reivindicaciones 1 a 25, para validar uno o más biomarcadores.
28. Uso de un método según una cualquiera de las reivindicaciones 1 a 25 como un método de diagnóstico.
Patentes similares o relacionadas:
Métodos y composiciones para el diagnóstico y pronóstico de lesión renal e insuficiencia renal, del 29 de Julio de 2020, de Astute Medical, Inc: Un método para evaluar el estado renal en un sujeto, que comprende: realizar una pluralidad de ensayos configurados para detectar una […]
Neuregulina para tratar la insuficiencia cardíaca, del 29 de Julio de 2020, de Zensun (Shanghai) Science & Technology, Co., Ltd: Neuregulina para usar en un método para tratar la insuficiencia cardíaca crónica en un paciente, donde el paciente tiene un nivel plasmático de NT-proBNP […]
Inmunomoduladores, del 29 de Julio de 2020, de BRISTOL-MYERS SQUIBB COMPANY: Un compuesto de la fórmula (I) **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en donde: A se selecciona de **(Ver fórmula)** en donde: […]
Detección de interacciones proteína a proteína, del 15 de Julio de 2020, de THE GOVERNING COUNCIL OF THE UNIVERSITY OF TORONTO: Un método para medir cuantitativamente la fuerza y la afinidad de una interacción entre una primera proteína de membrana o parte de la misma y una […]
Método para llevar a cabo el seguimiento de la enfermedad de Gaucher, del 15 de Julio de 2020, de Centogene GmbH: Un método para determinar la evolución de la enfermedad de Gaucher en un sujeto, que comprende la etapa de determinar en varios puntos en el […]
Procedimiento para evaluación de la función hepática y el flujo sanguíneo portal, del 15 de Julio de 2020, de The Regents of the University of Colorado, a body corporate: Procedimiento in vitro para la estimación del flujo sanguíneo portal en un individuo a partir de una única muestra de sangre o suero, comprendiendo el procedimiento: […]
Biomarcadores de pronóstico y predictivos y aplicaciones biológicas de los mismos, del 1 de Julio de 2020, de INSTITUT GUSTAVE ROUSSY: Un método para evaluar la sensibilidad o la resistencia de un tumor frente a un agente antitumoral, que comprende evaluar la cantidad de complejo eiF4E-eiF4G (complejo Cap-ON) […]
Evaluación asistida del pronóstico en la enfermedad inflamatoria, del 24 de Junio de 2020, de KINGS COLLEGE LONDON: Un método in vitro de recopilación de información útil para predecir el resultado clínico en un sujeto, en donde el sujeto tiene o se sospecha que tiene una enfermedad […]