COMPUESTOS BIFENILICOS COMO AGONISTAS SELECTIVOS PARA RECEPTORES GAMMA-RAR.
Compuestos bifenílicos de fórmula (I) **(Ver fórmula)** donde n es un número entero comprendido entre 1 y 3 inclusive,
así como las sales de los compuestos de fórmula (I)
Tipo: Resumen de patente/invención. Número de Solicitud: W06068976EP.
Solicitante: GALDERMA RESEARCH & DEVELOPMENT.
Nacionalidad solicitante: Francia.
Dirección: LES TEMPLIERS 2400 ROUTE DES COLLES,06410 BIOT.
Inventor/es: BIADATTI, THIBAUD, THOREAU, ETIENNE.
Fecha de Publicación: .
Fecha Concesión Europea: 9 de Septiembre de 2009.
Clasificación PCT:
- A61K31/192 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › que tienen grupos aromáticos, p. ej. sulindac, ácidos 2-aril-propiónicos, ácido etacrínico.
- A61P17/00 A61 […] › A61P ACTIVIDAD TERAPEUTICA ESPECIFICA DE COMPUESTOS QUIMICOS O DE PREPARACIONES MEDICINALES. › Medicamentos para el tratamiento de problemas dermatológicos.
- A61P17/06 A61P […] › A61P 17/00 Medicamentos para el tratamiento de problemas dermatológicos. › para el tratamiento de la psoriasis.
- A61P17/10 A61P 17/00 […] › Preparados contra el acné.
- C07C65/26 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07C COMPUESTOS ACICLICOS O CARBOCICLICOS (compuestos macromoleculares C08; producción de compuestos orgánicos por electrolisiso electroforesis C25B 3/00, C25B 7/00). › C07C 65/00 Compuestos que tienen grupos carboxilo unidos a átomos de carbono de ciclos aromáticos de seis miembros y que tienen uno de los grupos OH, O-metal,—CHO, cetona, éter, grupos, grupos, o grupos. › que contienen ciclos distintos a los ciclos aromáticos de seis miembros.
Fragmento de la descripción:
Compuestos bifenílicos como agonistas selectivos para receptores gamma-RAR.
La presente invención se relaciona con la utilización en terapia, especialmente en el campo de la dermatología, de compuestos bifenílicos substituidos por un radical aromático con actividad selectiva para el subtipo gamma de la familia de los receptores RAR.
Se describió una familia de compuestos bifenílicos en la solicitud de patente WO 99/10308. Estos compuestos son descritos como poseedores de una aplicación en el tratamiento tópico y sistémico de las afecciones dermatológicas ligadas a un trastorno de la queratinización y de las afecciones oftalmológicas especialmente.
La actividad de estos compuestos ha sido especialmente puesta en evidencia por pruebas de diferenciación de las células F9 de teratocarcinoma embrionario del ratón y pruebas de diferenciación de los queratinocitos en el hombre.
Por el contrario, este documento no tiene en cuenta en absoluto una eventual actividad específica de los compuestos frente al subtipo gamma de los receptores RAR.
En efecto, el subtipo gamma de la familia de los receptores RAR es con mucho mayoritario en la epidermis, donde representa aproximadamente un 90% del total de los receptores ("Retinoic acid receptors and binding proteins in human skin", Elder JT, Astrom A, Pettersson U, Tavakkol A, Krust A, Kastner P, Chambon P, Voorhees JJ: J. Invest. Dermatol. 1992; 98 (6 Supl.): 36S-41S; o: "Retinoic acid receptor expression in human skin keratinocytes and dermal fibroblasts in vitro", Redfern CP, Todd C. J. Cell Sci. 1992; 102 (Pt. 1): 113-21) y es la interacción con este receptor RAR gamma la responsable de la eficacia de los retinoides sobre la epidermis ("Retinoic acid receptor gamma mediates topical retinoid efficacy and irritation in animal models", Chen S, Ostrowski J, Whiting G, Roalsvig T, Hammer L, Currier SJ, Honeyman J, Kwasniewski B, Yu KL, Sterzycki R y col. J. Invest. Dermatol. 1995; 104 (5): 779-83).
La patente WO 2005/056516 divulga compuestos bifenílicos que actúan específicamente sobre los receptores RAR-gamma. Estos compuestos son utilizados en las enfermedades dermatológicas que implican a estos receptores.
Los receptores RAR gamma son, pues, el único objetivo en el tratamiento de patologías a nivel de la epidermis, como por ejemplo para el acné o la psoriasis o cualquier otra patología cutánea tratada mediante los retinoides.
Por otra parte, se pueden evitar ciertos efectos secundarios propios de los RAR alfa o RAR beta si se utilizan compuestos que tienen una acción selectiva sobre RAR gamma.
Sorprendentemente, se ha mostrado ahora que los compuestos según la invención presentan una actividad agonista selectiva para el subtipo gamma de la familia de los receptores RAR extremadamente interesante.
Los compuestos según la invención, agonistas selectivos del subtipo RAR gamma, permiten así prevenir y/o tratar diversas patologías o trastornos dermatológicos, disminuyendo los efectos secundarios habitualmente debidos a la acción de los principios activos sobre los subtipos RAR alfa y beta.
La presente invención tiene, pues, por primer objeto compuestos que pueden ser representados por la fórmula general siguiente:
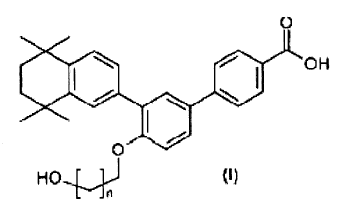
donde n es un número entero comprendido entre 1 y 3 inclusive, por lo tanto igual a 1, 2 ó 3, así como las sales de los compuestos de fórmula (I).
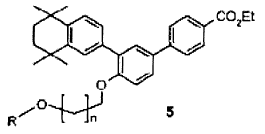
Según una forma de realización preferida, n es igual a 2 ó 3 y los compuestos de fórmula (I) preferidos son seleccionados entre el ácido 4'-(3-hidroxipropoxi)-3'-(5,5,8,8-tetrametil-5,6,7,8-tetrahidronaftalen-2-il)bifenil-4-carboxílico y el ácido 4'-(4-hidroxibutoxi)-3'-(5,5,8,8-tetrametil-5,6,7,8-tetrahidronaftalen-2-il)-bifenil-4-carboxílico.
Por sal farmacéuticamente aceptable, se entiende especialmente una sal de metal alcalino o una sal alcalinotérrea, o una sal de amina orgánica.
La invención contempla igualmente la utilización de al menos un compuesto de fórmula (I) para la preparación de una composición farmacéutica o cosmética destinada a prevenir y/o tratar patologías para las cuales se desea una actividad agonista selectiva para el subtipo gamma de la familia de los receptores RAR.
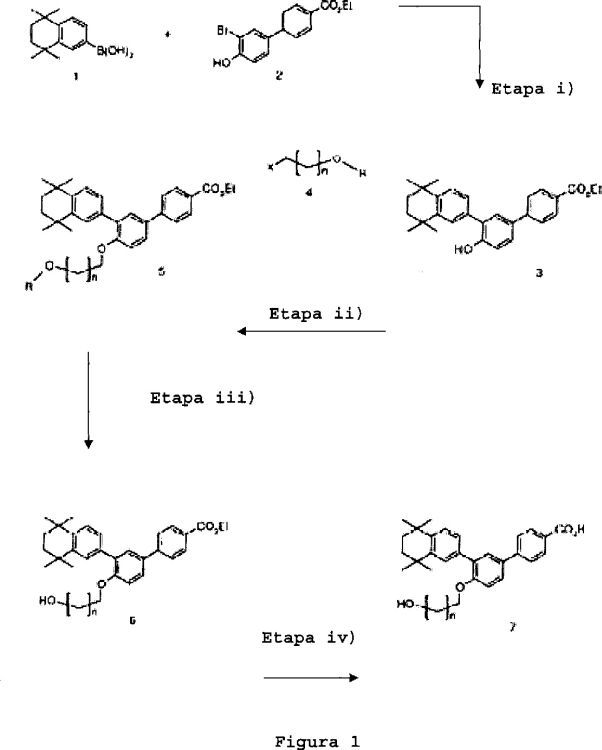
Se representa una vía de síntesis general para preparar los compuestos de fórmula (I) en el esquema según la figura 1.
Las materias primas y/o los reactivos utilizados son de disponibilidad comercial y/o pueden ser preparados según métodos conocidos de la literatura.
Según otro aspecto, la presente invención se relaciona igualmente con un procedimiento de preparación de los compuestos de fórmula (I) antes descritos consistente en las etapas siguientes:
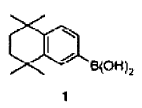
i) reacción de copulación, por ejemplo de tipo copulación de Suzuki entre el compuesto de fórmula 1 preparado por ejemplo como se describe en la solicitud de patente WO 99/10308:
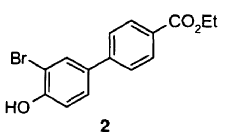
y el compuesto de fórmula 2 preparado por ejemplo como se describe en la solicitud de patente WO 99/10308:

para dar lugar al compuesto de fórmula 3,
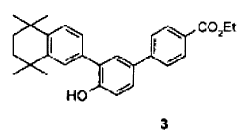
ii) reacción de eterificación, por ejemplo del tipo eterificación de Williamson o similar (véase, por ejemplo, Lerman, L y col.; Synthesis 2004, (18), 3043-3046), del compuesto de fórmula 3 con el compuesto de fórmula 4,
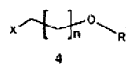
donde X representa un grupo saliente que permite una substitución nucleofílica, como por ejemplo un halógeno (preferiblemente un átomo de yodo o de bromo); R representa un hidrógeno o un grupo protector apropiado, tal como se describe en "Protective groups in organic synthesis" (Greene & Wuts, Wiley-Interscience 1991), por ejemplo acetilo o dimetil-terc-butilsililo, y n es un número entero comprendido entre 1 y 3, preferiblemente 2 ó 3, para dar lugar al compuesto de fórmula 5,
iii) en el caso de que R sea diferente de H, esta reacción ii) va seguida de una reacción de desprotección de la función alcohol del compuesto de fórmula 5 para dar lugar al compuesto de fórmula 6,

iv) saponificación de la función éster del compuesto obtenido en la etapa anterior, es decir, en la etapa ii) para los casos en que R=H o en la etapa iii) para los casos en que R es diferente de H, para dar lugar al compuesto de fórmula (I) correspondiente (compuesto 7 en la figura 1).
La etapa i) puede ser, por ejemplo, realizada en presencia de carbonato de potasio, de tetrakis(trifenilfosfino)paladio en una solución de tolueno.
La etapa ii) puede ser, por ejemplo, realizada en presencia de carbonato de cesio y de dimetilformamida y eventualmente de yoduro de potasio.
La etapa iii) puede ser realizada conforme a las reacciones de desprotección descritas en "Protective groups in organic synthesis" (Greene & Wuts, Wiley-Interscience 1991).
La etapa iv) puede ser, por ejemplo, realizada en presencia de hidróxido de sodio y de THF.
La presente invención tiene igualmente por objeto los compuestos de fórmula (I) tales como los antes descritos a modo de medicamento.
Según otro aspecto, la invención tiene por objeto una composición farmacéutica o cosmética caracterizada por incluir, en un vehículo farmacéutica o cosméticamente aceptable, al menos un compuesto de fórmula (I).
Por "vehículo farmacéutica o cosméticamente aceptable", se entiende un vehículo adaptado para una utilización en contacto con células humanas y de animales, sin toxicidad, irritación, respuesta alérgica indebida...
Reivindicaciones:
1. Compuestos bifenílicos de fórmula (I)
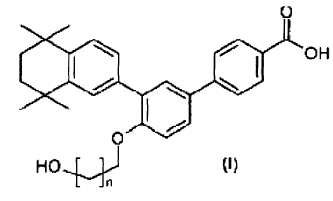
donde n es un número entero comprendido entre 1 y 3 inclusive, así como las sales de los compuestos de fórmula (I).
2. Compuesto según la reivindicación 1, caracterizado por ser seleccionado entre uno de los compuestos siguientes:
- el ácido 4'-(3-hidroxipropoxi)-3'-(5,5,8,8-tetrametil-5,6,7,8-tetrahidronaftalen-2-il)bifenil-4-carboxílico y
- el ácido 4'-(4-hidroxibutoxi)-3'-(5,5,8,8-tetrametil-5,6,7,8-tetrahidronaftalen-2-il)bifenil-4-carboxílico.
3. Compuesto según una cualquiera de las reivindicaciones precedentes a modo de medicamento.
4. Composición farmacéutica o cosmética, caracterizada por incluir en un vehículo farmacéutica o cosméticamente aceptable al menos un compuesto de fórmula (I) según una de las reivindicaciones 1 ó 2.
5. Composición farmacéutica o cosmética según la reivindicación 4, caracterizada por estar en forma adaptada para una administración por vía tópica.
6. Composición según la reivindicación 5, caracterizada por estar comprendida la cantidad de compuesto de fórmula (I) entre el 0,001% y el 3% en peso con respecto al peso total de la composición.
7. Utilización de un compuesto de fórmula (I) tal como se define en una de las reivindicaciones 1 ó 2 para la preparación de una composición farmacéutica destinada a la prevención y/o al tratamiento de patologías ligadas a una deficiencia de la activación del receptor RAR gamma.
8. Utilización según la reivindicación 7 para el tratamiento de una patología ligada a los trastornos de la diferenciación y/o de la proliferación celular.
9. Utilización según una de las reivindicaciones 7 ó 8 para el tratamiento del acné.
10. Utilización según una de las reivindicaciones 7 ó 8 para el tratamiento de la psoriasis.
11. Procedimiento de preparación de un compuesto de fórmula (I) según una de las reivindicaciones 1 ó 2, que consiste en las siguientes etapas:
i) la reacción de Suzuki entre el compuesto de fórmula 1

y el compuesto de fórmula 2
para dar lugar al compuesto de fórmula 3,
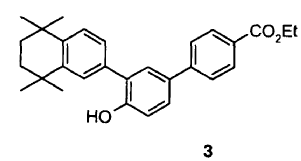
ii) eterificación del compuesto de fórmula 3 con el compuesto de fórmula 4,
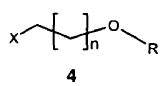
donde X representa un grupo saliente que permite una substitución nucleofílica, R representa un hidrógeno o un grupo protector y n es un número entero comprendido entre 1 y 3, para dar lugar al compuesto de fórmula 5,

iv) la saponificación de la función éster del compuesto obtenido en la etapa anterior.
12. Procedimiento de preparación de un compuesto de fórmula (I) según la reivindicación 11, caracterizado por incluir entre las etapas ii) y iv), en el caso en el cual el grupo R es diferente de H, una etapa iii) de desprotección de la función alcohol del compuesto de fórmula 5 para dar lugar al compuesto de fórmula 6:
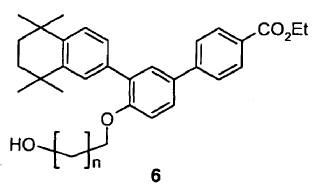
13. Procedimiento según una de las reivindicaciones 11 ó 12, caracterizado por ser X un halógeno.
14. Procedimiento según una de las reivindicaciones 11 a 13, caracterizado por ser R un grupo protector seleccionado entre acetilo y dimetil-terc-butilsililo.
Patentes similares o relacionadas:
Derivados de benzoxazinona para el tratamiento de enfermedades cutáneas, del 29 de Julio de 2020, de Sixera Pharma AB: Un compuesto según la Fórmula I **(Ver fórmula)** en donde R es -S-CH3 o -Cl, o una sal farmacéuticamente aceptable del mismo.
Composiciones farmacéuticas para uso tópico basadas en ácido hialurónico sulfatado como promotor de absorción de la piel, del 1 de Julio de 2020, de FIDIA FARMACEUTICI S.P.A.: Una composición farmacéutica para uso tópico que contiene ácido hialurónico sulfatado como promotor de absorción de la piel de diclofenaco, ketoprofeno, ibuprofeno […]
Composiciones tópicas que comprenden un corticosteroide y un retinoide para tratar la psoriasis, del 1 de Julio de 2020, de Bausch Health Ireland Limited: Una composición farmacéutica tópica para usar en el tratamiento de la psoriasis, la composición que comprende: (a) propionato de halobetasol […]
Derivado de amina cíclica y uso farmacéutico del mismo, del 1 de Julio de 2020, de TORAY INDUSTRIES, INC.: Un derivado de amina cíclica representado por la siguiente fórmula general (I): **(Ver fórmula)** donde R1 representa un grupo alquiloxi que tiene de 1 a 3 átomos […]
Sales de butirato para uso en enfermedades inflamatorias, del 17 de Junio de 2020, de Birrbeheer B.V: Una preparación que comprende una sal de butirato para uso en el tratamiento de un sujeto que padece una enfermedad que está asociada con inflamación sistémica, […]
Amidas heterocíclicas como inhibidores de quinasa, del 3 de Junio de 2020, de GlaxoSmithKline Intellectual Property Development Limited: Un compuesto que es **(Ver fórmula)** o un tautómero del mismo o una sal del mismo.
Composición farmacéutica tópica a base de alcanos semifluorados, del 13 de Mayo de 2020, de NOVALIQ GMBH: Una composición farmacéutica tópica para usar en la prevención o el tratamiento de una enfermedad o afección que afecta la piel o un apéndice de la piel, que comprende […]
Purina dionas como moduladores de la ruta de Wnt, del 29 de Abril de 2020, de AGENCY FOR SCIENCE, TECHNOLOGY AND RESEARCH: Un compuesto que tiene la estructura (I) **(Ver fórmula)** o una sal del mismo; en el que: R1, R2, R3, R4 y R5 son cada uno, independientemente, […]