SISTEMA PARA LA LIBERACIÓN CONTROLADA DE MORFINA.
Una composición farmacéutica para la liberación controlada de al menos un opioide en un medio acuoso por erosión de al menos una superficie de la composición,
donde la composición comprende i) una composición matricial que contiene a) un polímero o una mezcla de polímeros, donde el polímero comprende un polietilenglicol, un óxido de polietileno y/o un copolímero de bloques de óxido de etileno y óxido de propileno, b) un opioide como principio activo y, opcionalmente, c) uno o más excipientes farmacéuticamente aceptables, y ii) un recubrimiento que tiene al menos un orificio en exposición en la una superficie de dicha matriz, donde el recubrimiento contiene a) un primer derivado de celulosa que tiene propiedades termoplásticas y que es considerablemente insoluble en el medio acuoso en el cual se va a usar la composición, b) y al menos uno de b) un segundo derivado de celulosa que es soluble o dispersable en agua, c) un plastificante, y d) un relleno, donde la composición matricial tiene forma semejante a un cono de modo que el área superficial expuesta al medio acuoso aumenta al menos durante la erosión inicial de la composición matricial; y la disolución del opioide - cuando se analiza en una prueba de disolución de conformidad con la USP 24, NF 19, (711), Disolución, aparato 2 equipado con una paleta; con o sin la aplicación de dispositivos de inmersión produce una liberación de orden cero de al menos 80% del opioide contenido en la composición, y aproximadamente 75% p/p del opioide se libera desde la composición en 4 a 10 horas
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/DK2004/000215.
Solicitante: Egalet Ltd.
Nacionalidad solicitante: Reino Unido.
Dirección: Egmont House 8 Clifford Street London W1S 2LQ REINO UNIDO.
Inventor/es: BAR-SHALOM, DANIEL, FISCHER,GINA, SLOT,LILLIAN, ANDERSEN,CHRISTINE.
Fecha de Publicación: .
Fecha Solicitud PCT: 26 de Marzo de 2004.
Clasificación Internacional de Patentes:
- A61K31/485 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › Derivados del morfinano, p. ej. morfina, codeína.
- A61K9/00Z4
Clasificación PCT:
- A61K9/22 A61K […] › A61K 9/00 Preparaciones medicinales caracterizadas por un aspecto particular. › del tipo de liberación prolongada o discontinua.
Clasificación antigua:
- A61K9/22 A61K 9/00 […] › del tipo de liberación prolongada o discontinua.
Países PCT: Austria, Bélgica, Suiza, Alemania, Dinamarca, España, Francia, Reino Unido, Grecia, Italia, Liechtensein, Luxemburgo, Países Bajos, Suecia, Mónaco, Portugal, Irlanda, Eslovenia, Finlandia, Rumania, Chipre, Lituania, Letonia, Ex República Yugoslava de Macedonia, Albania.
PDF original: ES-2360102_T3.pdf




Fragmento de la descripción:
45
55
Campo de la invención
La presente invención se refiere a una nueva composición farmacéutica para la liberación controlada de un opioide, como por ejemplo morfina, en un medio acuoso. La composición farmacéutica es una composición matricial recubierta en la cual la composición matricial comprende a) un polímero o una mezcla de polímeros, b) un opioide como por ejemplo morfina y, opcionalmente, c) uno o más excipientes farmacéuticamente aceptables. El recubrimiento permanece intacto durante la fase de liberación y después de eso puede desmenuzarse y/o erosionarse. Además, el recubrimiento cubre la composición matricial de modo que solo un área superficial específica de la composición matricial está sujeta a erosión en un medio acuoso, es decir, el área superficial de la cual se libera el principio activo se mantiene considerablemente constante durante el lapso de tiempo.
El polímero mencionado en a) antes, puede ser adecuadamente un polímero considerablemente soluble en agua
o cristalino o una mezcla de polímeros considerablemente solubles en agua y/o cristalinos.
El diseño de la composición farmacéutica se basa en el descubrimiento de que es posible controlar la liberación desde dicha composición asegurando que la liberación tenga lugar predominantemente por erosión. Para asegurar una liberación basada en la erosión se debe obtener un equilibrio entre la velocidad de difusión de agua dentro de la composición matricial y la velocidad de disolución de la composición matricial.
En consecuencia, la invención se refiere a una composición farmacéutica para uso oral que contiene un opioide, que proporciona una liberación de orden cero basada en controlar el equilibrio entre la velocidad de erosión de la matriz y la velocidad de difusión dentro de la matriz.
La invención también proporciona varias combinaciones de composiciones opioides de liberación inmediata y composiciones opioides de liberación controlada.
Divulgación de la invención
La presente invención se refiere a una composición de liberación controlada que contiene un opioide como principio terapéutica, profiláctica y/o diagnósticamente activo. Los opioides se usan generalmente en tratamiento analgésico y existe la necesidad de desarrollar composiciones para uso oral, que tengan una menor frecuencia de administración. Por lo tnato, se preferirían administraciones una o dos veces al día. Una composición de liberación controlada de acuerdo con la invención apunta a una liberación de orden cero del opioide en un patrón predeterminado para reducir y/o retrasar la concentración plasmática máxima sin afectar la magnitud de la biodisponibilidad del fármaco. La frecuencia de efectos secundarios indeseados se puede reducir, y debido al retraso en el tiempo que puede llevar obtener la concentración plasmática máxima y la prolongación del tiempo en la concentración plasmática terapéuticamente activa, la frecuencia de la administración se puede reducir a una dosis que se tome solo una o dos veces al día. Esto también sirve mejorar la obediencia del paciente. Otra ventaja de la composición de liberación controlada es que se evitan concentraciones locales elevadas del opioide en el tubo digestivo debido al mecanismo de liberación basado en la erosión.
Los pacientes que sufren de dolores crónicos muy a menudo requieren una dosis diaria alta de un analgésico. Si dicha dosis alta de un opioide debe ser administrada solo una o dos veces al día, la liberación desde la composición debe ser segura. La composición debe ser también estable en el almacenamiento con respecto a su estabilidad química y física.
La morfina es probablemente uno de los analgésicos más comúnmente utilizados en el control del dolor intenso. Debido a la corta semivida de la morfina de aproximadamente 2 horas, el analgésico se debe administrar frecuentemente, por ejemplo cada 3 a 4 horas, para mantener al paciente sin dolor. Este régimen puede requerir una dosis a las 2 a.m. para evitar la reaparición del dolor en la madrugada.
Para obtener concentraciones plasmáticas terapéuticas con una administración menos frecuente, los analgésicos de liberación controlada eficaces están en continuo desarrollo. Un analgésico de liberación controlada ideal debería exhibir las propiedades siguientes: prevenir el dolor "intercurrente", eliminar las principales fluctuaciones de la concentración plasmática, prolongar los niveles eficaces del fármaco, producir una analgesia efectiva sin efectos secundarios indeseados como la sedación. [0010] En otras palabras, además de una administración menos frecuente, el verdadero desafío en la administración de liberación controlada se puede expresar mediante el objetivo de disminuir la incidencia de efectos adversos y al mismo tiempo aumentar el efecto del tratamiento. Esto solo se puede obtener mediante la interacción entre las propiedades farmacológicas específicas del principio activo y la composición.
Muchos productos de liberación controlada del mercado carecen de una verdadera liberación controlada. De hecho, Dolcontin®, que cubre la mayor parte del mercado de la morfina de liberación sostenida, tiene un perfil plasmático bastante similar al de una formulación de liberación inmediata de la dosis, solo con una ligera disminución de la absorción rápida inicial. Durante la administración repetida, la concentración plasmática con este producto presentará necesariamente los máximos y mínimos indeseados como también se muestra en la figura 18 que compara la formulación comercial MS Contin de dos veces al día con la formulación de múltiples gránulos Kadian, en la que ambas demuestran una fluctuación durante el día donde la concentración mínima es menos de la mitad de la concentración máxima para cada una de las dos formulaciones. Dicho grado de fluctuación en la concentración durante el intervalo de dosificación puede ser evitado con la composición opioide de acuerdo con la presente invención.
Liberación controlada no es solo el efecto de prolongar la liberación del principio activo desde la composición aumentando la dosis y disminuyendo la absorción rápida inicial. El efecto optimizado solo se obtiene cuando se logra el equilibrio adecuado entre la concentración máxima, el tiempo durante el cual la concentración plasmática está por encima del nivel terapéutico mínimo y la dosis administrada.
Altas concentraciones o un aumento rápido en la concentración de morfina son factores importantes que dan lugar a efectos secundarios entre ellos el riesgo de volverse adicto a la morfina. El temor a la adicción es a menudo el principal obstáculo para la iniciación del tratamiento para el dolor con morfina, por lo demás eficaz, tanto desde el punto de vista del personal clínico como de los mismos pacientes.
También se deben evitar concentraciones altas durante períodos más largos porque inducen resistencia en el nivel del receptor. Mediante "altas" se quiere dar a entender cualquier concentración plasmática por encima del nivel de alivio del dolor. Otros efectos secundarios importantes de la morfina son el efecto de depresión respiratoria y sedación; ambos están muy correlacionados con la concentración plasmática. Además, el control de la memoria y motor, aspectos importantes en relación con el tratamiento a largo plazo, también pueden ya estar presentes dentro del nivel terapéutico y en consecuencia, se debe evitar cualquier concentración innecesariamente alta de morfina.
De acuerdo con la presente invención es posible obtener una composición, que sea eficaz para tratar el dolor y que simultáneamente tenga una o más de las ventajas siguientes: una concentración sérica máxima del opioide relativamente baja y efecto de alivio del dolor, concentraciones séricas promedio relativamente disminuidas pero aún con un efecto de alivio del dolor, concentraciones mínimas de opioide pero aún con un efecto de alivio del dolor. Cada uno de esos factores se puede asociar con un riesgo menor de efectos secundarios. Esta conclusión debe ser respaldada por informes menos frecuentes de efectos secundarios por parte de los pacientes que reciben dicha composición en un estudio clínico que compara composiciones de liberación controlada que tienen un perfil de disolución que no es de orden cero. Los eventos adversos que se van a informar en dicho estudio incluyen sedación, náuseas, mareos, vértigo, estreñimiento crónico, retención de orina, prurito,... [Seguir leyendo]
Reivindicaciones:
1. Una composición farmacéutica para la liberación controlada de al menos un opioide en un medio acuoso por erosión
de al menos una superficie de la composición, donde la composición comprende i) una composición matricial que contiene a) un polímero o una mezcla de polímeros, donde el polímero comprende un polietilenglicol, un óxido de polietileno y/o un copolímero de bloques de óxido de etileno y óxido de propileno, b) un opioide como principio activo y, opcionalmente, c) uno o más excipientes farmacéuticamente aceptables, y ii) un recubrimiento que tiene al menos un orificio en exposición en la una superficie de dicha matriz, donde el recubrimiento contiene
a) un primer derivado de celulosa que tiene propiedades termoplásticas y que es considerablemente
insoluble en el medio acuoso en el cual se va a usar la composición, b) y al menos uno de b) un segundo derivado de celulosa que es soluble o dispersable en agua, c) un plastificante, y d) un relleno, donde la composición matricial tiene forma semejante a un cono de modo que el área superficial expuesta al medio acuoso aumenta al menos durante la erosión inicial de la composición matricial; y la disolución del opioide - cuando se analiza en una prueba de disolución de conformidad con la USP 24, NF 19, (711), Disolución, aparato 2 equipado con una paleta; con o sin la aplicación de dispositivos de inmersión produce una liberación de orden cero de al menos 80% del opioide contenido en la composición, y aproximadamente 75% p/p del opioide se libera desde la composición en 4 a 10 horas.
2. Una composición farmacéutica de acuerdo con la reivindicación 1, en la que el área superficial expuesta aumenta durante la primera 0.5 hora tal como, por ejemplo, durante la primera 1 hora, durante las primeras 1.5 horas, durante las primeras 2 horas, durante las primeras 3 horas, durante las primeras 5 horas o durante las primeras 6 horas.
3. Una composición farmacéutica de acuerdo con la reivindicación 1 o 2, en la que el aumento en el área superficial se relaciona con un aumento del diámetro del área superficial de al menos un área superficial expuesta en el momento de la erosión de esa superficie, y la relación entre el diámetro mayor y el menor disminuye entre aproximadamente 2.5 y 1 durante la erosión, tal como entre aproximadamente 2 y 1, tal como entre aproximadamente 1.8 y 1, tal como entre aproximadamente 1.6 y 1, tal como entre aproximadamente 1.5 y 1, tal como entre aproximadamente 1.4 y 1 tal como entre aproximadamente 1.3 y 1, tal como entre aproximadamente 1.2 y 1.
4. Una composición farmacéutica de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la concentración plasmática media 8 horas después de la administración oral a al menos 6 humanos adultos sanos es al menos 40% de la concentración máxima media obtenida por la dosis, como, por ejemplo, al menos 50% o al menos 60% de la concentración máxima media.
5. Una composición farmacéutica de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la concentración plasmática media 10 horas después de la administración oral a al menos 6 humanos adultos sanos es al menos 35% de la concentración máxima media obtenida por la dosis, tal como, por ejemplo, al menos 40% o al menos 50% de la concentración máxima media.
6. Una composición farmacéutica de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la concentración plasmática media 12 horas después de la administración oral a al menos 6 humanos adultos sanos es al menos 25% de la concentración máxima media obtenida por la dosis, tal como, por ejemplo, al menos 30%, al menos 35%, al menos 40% o al menos aproximadamente 45% de la concentración máxima media.
7. Una composición farmacéutica de acuerdo con cualquiera de las reivindicaciones precedentes para la administración una o dos veces al día.
8. Una composición farmacéutica de acuerdo con cualquiera de las reivindicaciones precedentes para la administración una vez al día.
9. Una composición farmacéutica de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la concentración plasmática media después de la administración oral de una dosis única a al menos 6 humanos adultos sanos es al menos 33% de la concentración máxima media durante al menos 15 horas tal como, por ejemplo, durante al menos 17 horas, durante al menos 19 horas o durante al menos 20 horas.
10. Una composición farmacéutica de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la concentración plasmática media después de la administración oral de una dosis única a al menos 6 humanos adultos sanos es al menos 50% de la concentración máxima media durante al menos 6 horas tal como, por ejemplo, durante al menos 8 horas, durante al menos 9 horas, durante al menos 10 horas o durante al menos 11 horas.
11. Una composición farmacéutica de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la
concentración plasmática media después de la administración oral de una dosis única a al menos 6 humanos adultos sanos es al menos 75% de la concentración máxima media durante al menos 3 horas tal como, por ejemplo, durante al menos 3.3 horas, durante al menos 3.5 horas, durante al menos 3.7 horas o durante al menos 3.9 horas.
12. Una composición farmacéutica de acuerdo con la reivindicación 8, en la que la concentración plasmática media 12 horas después de la administración oral de una dosis única es al menos 20% tal como, por ejemplo, al menos 25% o al menos 30% de la concentración máxima media, y/o la concentración plasmática media 18 horas después de la administración oral es al menos 20% tal como, por ejemplo, al menos 25%, al menos 30% o al menos 35% de la concentración máxima media, y/o la concentración plasmática media 24 horas después de la administración oral es al menos 20% tal como, por ejemplo, al menos 25% o al menos aproximadamente 30% de la concentración máxima media.
13. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que el opioide se selecciona del grupo que consiste en alfentanilo, alilprodina, alfaprodina, anileridina, bencilmorfina, becitramida, buprenorfina, butorfanol, clonitaceno, codeína, ciclazocina, desomorfina, dextromoramida, dezocina, diampromida, dihidrocodeína, dihidromorfina, dimenoxadol, dimefeptanol, dimetiltiambuteno, butirato de dioxafetilo, dipipanona, eptazocina, etoheptacina, etilmetiltiambuteno, etilmorfina, etonitaceno, fentanilo, heroína, hidrocodona, hidromorfona, hidroxipetidina, isometadona, dextropropoxifeno, cetobemidona, levalorfano, levorfanol, levofenacilmorfano, lofentanilo, meperidina, meptacinol, metazocina, metadona, metopón, morfina, morfina-6-glucurónido, morfina-3-glucurónido, mirofina, nalbufina, narceína, nicomorfina, norlevorfanol, normetadona, nalorfina, normorfina, norpipanona, opio, oxicodona, oximorfona, papaveretum, pentazocina, fenadoxona, fenomorfano, fenazocina, fenoperidina, piminodina, piritramida, proheptacina, promedol, properidina, propiram, propoxifeno, sufentanilo, tilidina, tramadol y sus sales complejos, solvatos o anhidratos farmacéuticamente aceptables; y mezclas de estos.
14. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que el opioide es morfina, morfina 6-glucurónido, morfina 3-glucurónido o sus mezclas.
15. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que el principio activo es un polvo farmacéuticamente activo.
16. Una composición de acuerdo con la reivindicación 15, en la que el polvo tiene un tamaño de partícula entre aproximadamente 0.1 µm y aproximadamente 500 µm, típicamente entre aproximadamente 0.5 µm y aproximadamente 300 µm, más típicamente entre aproximadamente 1 µm y aproximadamente 200 µm, especialmente entre aproximadamente 5 µm y aproximadamente 100 µm.
17. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que el opioide está presente en la composición matricial en una concentración entre aproximadamente 0.1 y aproximadamente 98% p/p tal como, por ejemplo a lo sumo aproximadamente 90% p/p, a lo sumo aproximadamente 85% p/p, a lo sumo aproximadamente 80% p/p, a lo sumo aproximadamente 75% p/p, a lo sumo aproximadamente 70% p/p, a lo sumo aproximadamente 65% p/p
o a lo sumo aproximadamente 60% p/p.
18. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que aproximadamente 50% p/p del opioide se libera desde la composición en 3 a 5 horas según se mide por la prueba de disolución descrita en este documento.
19. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la composición - cuando se analiza en un sistema de disolución in vitro de conformidad con la USP (paletas, 50 rpm, solución amortiguadora de pH 6.8 sin dispositivos de inmersión) - libera al menos aproximadamente 80% p/p de la cantidad total de opioide en un período entre aproximadamente 5 y aproximadamente 10 horas tal como, por ejemplo, entre aproximadamente 6 y aproximadamente 9 horas tal como, por ejemplo entre aproximadamente 7 y 8 horas o aproximadamente 7.5 horas después de comenzar la prueba.
20. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la composición - cuando se analiza en un sistema de disolución in vitro de conformidad con la USP (paletas, 50 rpm, solución amortiguadora de pH 6.8 con dispositivos de inmersión) - libera al menos aproximadamente 80% p/p de la cantidad total de opioide en un período entre aproximadamente 4 y aproximadamente 9 horas tal como, por ejemplo entre aproximadamente 5 y aproximadamente 8 horas tal como, por ejemplo, entre aproximadamente 6 y 7 horas o aproximadamente 6 horas después de comenzar la prueba.
21. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la composición - cuando se analiza en un sistema de disolución in vitro de conformidad con la USP (paleta, 50 rpm, solución amortiguadora de pH 6.8) - libera el opioide para que cuando se alcanza el 30% del tiempo necesario para liberar al menos aproximadamente 80% p/p de la cantidad total de opioide - entonces se libera entre aproximadamente 10% y aproximadamente 50% tal como, por ejemplo, entre aproximadamente 15% y aproximadamente 40% p/p, entre aproximadamente 20% y aproximadamente 30% o aproximadamente 23 a 27% p/p.
22. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la composición - cuando se analiza en un sistema de disolución in vitro de conformidad con la USP (paleta, 50 rpm, solución amortiguadora de pH 6.8) - libera el opioide para que cuando se alcanza el 50% del tiempo necesario para liberar al menos aproximadamente 80% p/p de la cantidad total del opioide - entonces se libera entre aproximadamente 20% y aproximadamente 60% tal como, por ejemplo, entre aproximadamente 30% y aproximadamente 50% p/p o aproximadamente 42 a 47% p/p.
23. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la composición - cuando se analiza en un sistema de disolución in vitro de conformidad con la USP (paleta, 50 rpm, solución amortiguadora de pH 6.8) - libera el opioide para que cuando se alcanza el 60% del tiempo necesario para liberar al menos aproximadamente 80% p/p de la cantidad total del opioide - entonces se libera entre aproximadamente 30% y aproximadamente 80% p/p tal como, por ejemplo, entre aproximadamente 40% y aproximadamente 70% p/p, entre aproximadamente 50 y aproximadamente 60% o aproximadamente 52 a 58% p/p.
24. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la composición - cuando se analiza en un sistema de disolución in vitro de conformidad con la USP (paleta, 50 rpm, solución amortiguadora de pH 6.8) - libera el opioide de la manera siguiente:
dentro de las primeras 2 horas después de comenzar la prueba se libera entre aproximadamente 0 y aproximadamente 30% p/p del opioide. dentro de las primeras 5 horas después de comenzar la prueba se libera entre aproximadamente 25% y aproximadamente 80% p/p del opioide. dentro de las primeras 7 horas después de comenzar la prueba se libera entre aproximadamente 40% y aproximadamente 100% p/p del opioide.
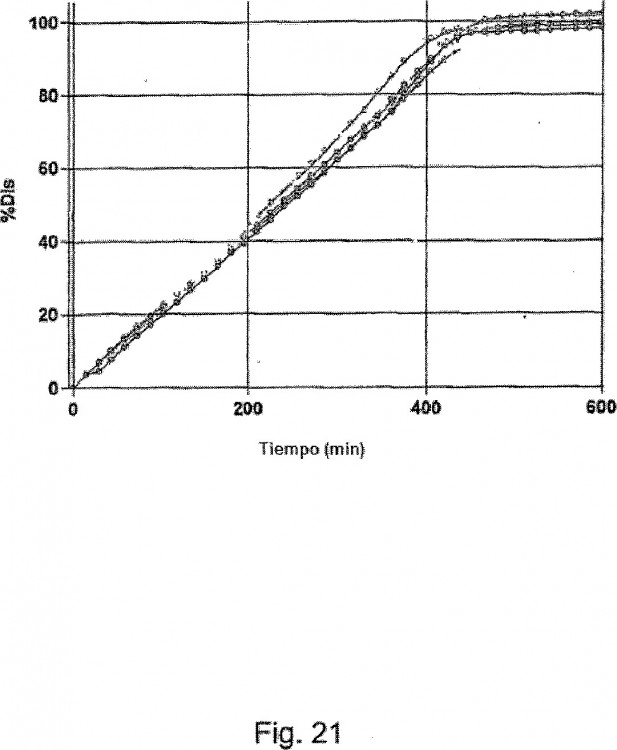
25. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la composición tiene un patrón de disolución que se asemeja al de la figura 21 de este documento, habiendo sido analizada la composición en condiciones similares.
26. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes en la que la composición matricial tiene una forma seleccionada entre las formas definidas en la tabla A de este documento.
27. Una composición de acuerdo con la reivindicación 23, en la que la forma corresponde a un cono 1 o 5 de la tabla A de este documento.
28. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que el óxido de polietileno tiene un peso molecular entre aproximadamente 20 000 Dalton, tal como, por ejemplo, entre aproximadamente 20 000 y aproximadamente 70 000 Dalton, entre aproximadamente 20 000 y aproximadamente 600 000 Dalton, entre aproximadamente 35 000 y aproximadamente 500 000 Dalton, entre aproximadamente 35 000 y aproximadamente 400 000 Dalton, entre aproximadamente 35 000 y aproximadamente 300 000 Dalton, entre aproximadamente 50 000 y aproximadamente 300 000 Dalton, tal como, por ejemplo aproximadamente 35 000 Dalton, aproximadamente 50 000 Dalton, aproximadamente 75 000 Dalton, aproximadamente 100 000 Dalton, aproximadamente 150 000 Dalton, aproximadamente 200 000 Dalton, aproximadamente 250 000 Dalton, aproximadamente 300 000 Dalton o aproximadamente 400 000 Dalton.
29. Una composición de acuerdo con cualquiera de las reivindicaciones 1 a 27, en la que el copolímero de bloques de óxido de etileno y óxido de propileno contiene hasta aproximadamente 30% p/p del bloque basado en óxido de propileno, y tiene un peso molecular de aproximadamente 5000 Dalton, típicamente aproximadamente 5000 a aproximadamente 30 000 Dalton tal como, por ejemplo entre aproximadamente 8000 y aproximadamente 15 000 Dalton.
30. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la matriz contiene un polímero que tiene un punto de fusión entre aproximadamente 20 y 120 °C tal como, por ejemplo entre aproximadamente 30 y aproximadamente 100 °C o entre aproximadamente 40 y aproximadamente 80 °C.
31. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que el polímero es un óxido de polietileno que tiene un peso molecular de al menos 100 000 Dalton y a lo sumo 300 000 Dalton.
32. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la composición matricial contiene PEO 200 000 NF y/o PEO 200 000 LF.
33. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la matriz contiene un excipiente farmacéuticamente aceptable.
34. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que el excipiente farmacéuticamente aceptable se selecciona del grupo que consiste en ácidos inorgánicos, bases inorgánicas, sales inorgánicas, ácidos o bases orgánicos y sus sales farmacéuticamente aceptables, sacáridos, oligosacáridos, polisacáridos, celulosa y derivados de celulosa.
35. Una composición de acuerdo con la reivindicación 34, donde el ácido orgánico es un ácido mono, di, oligo, policarboxílico o aminoácidos tales como, por ejemplo ácido acético, ácido etanoico, ácido succínico, ácido cítrico, ácido tartárico, ácido acrílico, ácido benzoico, ácido málico, ácido maleico, ácido adípico, ácido angélico, ácido ascórbico/vitamina C, ácido carbámico, ácido cinámico, ácido citramálico, ácido fórmico, ácido fumárico, ácido gálico, ácido gentísico, ácido glutacónico, ácido glutárico, ácido glicérico, ácido glicólico, ácido glioxílico, ácido láctico, ácido levulínico, ácido malónico, ácido mandélico, ácido oxálico, ácido oxámico, ácido pimélico, ácido pirúvico, ácido aspártico y ácido glutámico.
36. Una composición de acuerdo con la reivindicación 34, donde el ácido inorgánico es pirofosfórico, glicerofosfórico, fosfórico tal como orto o meta fosfórico, ácido bórico, ácido clorhídrico o ácido sulfúrico .
37. Una composición de acuerdo con la reivindicación 34, en la que los compuestos inorgánicos adecuados incluyen aluminio.
38. Una composición de acuerdo con la reivindicación 34, en la que las bases orgánicas adecuadas se seleccionan del grupo que consiste en p-nitrofenol, succinimida, bencenosulfonamida, 2-hidroxi-2-ciclohexenona, imidazol, pirrol, dietanolamina, etilenamina, tris (hidroximetil)aminometano, hidroxilamina y derivados de aminas, citrato de sodio, anilina e hidrazina.
39. Una composición de acuerdo con la reivindicación 34, en la que las bases inorgánicas adecuadas. se seleccionan del grupo que consiste en óxido de aluminio que incluye óxido de aluminio trihidrato, alúmina, hidróxido de sodio, hidróxido de potasio, carbonato de calcio, carbonato de amonio, hidróxido de amonio, KOH.
40. Una composición de acuerdo con la reivindicación 34, en la que la sal farmacéuticamente aceptable de un ácido inorgánico, es por ejemplo una sal de un metal alcalino o una sal de un metal alcalinotérreo incluidas fosfato de sodio, fosfato ácido de sodio, fosfato ácido disódico, fosfato de potasio, fosfato ácido de potasio, fosfato dibásico de potasio, fosfato de calcio, fosfato dicálcico, sulfato de sodio, sulfato de potasio, sulfato de calcio, carbonato de sodio, carbonato ácido de sodio, carbonato de potasio, carbonato ácido de potasio, carbonato de calcio, carbonato de magnesio, acetato de sodio, acetato de potasio, acetato de calcio, succinato de sodio, succinato de potasio, succinato de calcio, citrato de sodio, citrato de potasio, citrato de calcio, tartrato de sodio, tartrato de potasio, tartrato de calcio, gluconato de cinc, sulfato de cinc.
41. Una composición de acuerdo con la reivindicación 34, en la que la sal inorgánica es cloruro de sodio, cloruro de potasio, cloruro de calcio o cloruro de magnesio.
42. Una composición de acuerdo con la reivindicación 34, en la que el excipiente farmacéuticamente aceptable se selecciona entre glucosa y otros monosacáridos, ribosa, arabinosa, xilosa, lixosa, alosa, altrosa, inositol, glucosa, sorbitol, manosa, gulosa, idosa, galactosa, talosa, manitol, fructosa, lactosa, sacarosa, y otros disacáridos, dextrina, dextrano u otros polisacáridos, amilosa, xilano, celulosa y derivados de celulosa que incluyen celulosa microcristalina, metilcelulosa, etilcelulosa, etilhidroxietilcelulosa, etilmetilcelulosa, hidroxietilcelulosa, hidroxietilmetilcelulosa, carboximetilcelulosa, hidroxipropilcelulosa, hidroximetilpropilcelulosa, hidroxipropilmetilcelulosa, amilopectina, pectina, almidón, almidón sódico, caolín, bentonita, acacia, ácido algínico, alginato de sodio, alginato de calcio, gelatina, dextrosa, molasas, extracto de musgo de Irlanda, goma panwar, goma ghatti, mucílago de vaina de isapol, veegum, glicolato, estearato de magnesio, estearato de calcio, ácido esteárico, talco, dióxido de titanio, dióxido de silicio, arcillas, croscarmelosa, gomas y agar.
43. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes que contiene además un excipiente farmacéuticamente aceptable seleccionado del grupo que consiste en rellenos, diluyentes, desintegrantes, deslizantes, ajustadores del pH, ajustadores de la viscosidad, agentes para aumentar o disminuir la solubilidad, agentes osmóticamente activos y solventes.
44. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que al menos un opioide tiene una solubilidad de a lo sumo aproximadamente 3 mg/ ml tal como, por ejemplo a lo sumo aproximadamente 1 mg/ml, a lo sumo aproximadamente 0.1 mg/ml, a lo sumo aproximadamente 0.05 mg/ml tal como, por ejemplo a lo sumo aproximadamente 0.001 mg/ml en agua, a temperatura ambiente.
45. Una composición de acuerdo con la reivindicación 41, en la que la composición matricial contiene un excipiente farmacéuticamente aceptable que tiene una solubilidad de al menos 1 mg/ml tal como, por ejemplo al menos aproximadamente 3 mg/ml, al menos aproximadamente 5 mg/ml, al menos aproximadamente 10 mg/ml, al menos aproximadamente 25 mg/ml o al menos aproximadamente 50 mg/ml en agua, a temperatura ambiente.
46. Una composición de acuerdo con cualquiera de las reivindicaciones 1 a 43, en la que el al menos un opioide tiene una solubilidad de al menos aproximadamente 3 mg/ml tal como, por ejemplo al menos aproximadamente 5 mg/ml, al menos aproximadamente 10 mg/ml, al menos aproximadamente 20 mg/ml, al menos aproximadamente 50 mg/ml o al menos aproximadamente 100 mg/ml en agua, a temperatura ambiente.
47. Una composición de acuerdo con la reivindicación 46, en la que la composición matricial contiene un excipiente farmacéuticamente aceptable que tiene una solubilidad de a lo sumo aproximadamente 3 mg/ml/ ml tal como, por ejemplo a lo sumo aproximadamente 1 mg/ml, a lo sumo aproximadamente 0.1 mg/ml, a lo sumo aproximadamente 0.05 mg/ml tal como, por ejemplo a lo sumo aproximadamente 0.001 mg/ml en agua a temperatura ambiente.
48. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que en el medio acuoso en el cual se usa la composición, el recubrimiento no se desmenuza ni erosiona completamente antes de que la matriz se haya erosionado completamente.
49. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que dicho primer derivado de celulosa es un éter de celulosa al cual, cuando se calienta, se le puede dar forma por moldeo o extrusión, que incluye moldeo por inyección, moldeo por soplado y moldeo por compresión.
50. Una composición de acuerdo con la reivindicación 49 en la cual el éter de celulosa comprende al menos una etilcelulosa.
51. Una composición de acuerdo con cualquiera de las reivindicaciones 1 a 48 en la cual dicho primer derivado de celulosa se selecciona del grupo que consiste en acetato de celulosa, propionato de celulosa y nitrato de celulosa.
52. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes en la cual dicho segundo derivado de celulosa se selecciona del grupo que consiste en metilcelulosa, carboximetilcelulosa y sus sales, acetato ftalato de celulosa, celulosa microcristalina, etilhidroxietilcelulosa, etilmetilcelulosa, hidroxietilcelulosa, hidroxietilmetilcelulosa, hidroxipropilcelulosa, hidroximetilcelulosa e hidroximetilpropilcelulosa.
53. Una composición de acuerdo con la reivindicación 52 en la cual dicha sal de carboximetilcelulosa se selecciona del grupo que consiste en sales de metales alcalinos y alcalinotérreos.
54. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la cual dicho plastificante se selecciona del grupo que consiste en ésteres fosfato; ésteres ftalato; amidas; aceites minerales; ácidos grasos y sus ésteres con polietilenglicol, glicerina o azúcares; alcoholes grasos y sus éteres con polietilenglicol, glicerina o azúcares; aceites vegetales y aceites vegetales hidrogenados; nitrobenceno, disulfuro de carbono, salicilato de β-naftilo, glicolato de ftalilo y ftalato de diocilo.
55. Una composición de acuerdo con la reivindicación 54 en la cual dicho alcohol graso se selecciona del grupo que consiste en alcohol cetoestearílico, alcohol cetílico, alcohol estearílico, alcohol oleílico y alcohol miristílico.
56. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes en la cual dicho plastificante es un surfactante no iónico.
57. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que la composición matricial no contiene monoestearato de polietilenglicol 2000 ni monoestearato de polietilenglicol 400.
58. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, en la que está presente el excipiente farmacéuticamente aceptable y se selecciona entre manitol, xilitol, sorbitol e inositol.
59. Una composición de acuerdo con las reivindicaciones 1 a 58, en la que el excipiente farmacéuticamente aceptable es un óxido de aluminio.
60. Una composición de acuerdo con cualquiera de las reivindicaciones precedentes, que contiene PEO 200 000 como polímero y manitol y/o óxido de aluminio como excipiente farmacéuticamente aceptable.
61. El uso de una composición, como la definida en cualquiera de las reivindicaciones 1 a 60, para la fabricación de un medicamento para el tratamiento del dolor sensible a un opioide.
62. El uso de acuerdo con la reivindicación 61, donde la cantidad de opioide en una base diaria suficiente para tratar el dolor en el paciente es menor que la cantidad de opioide suficiente para tratar el dolor en una medida semejante mediante el uso de una composición de liberación inmediata.
63. El uso de acuerdo con la reivindicación 62, en que el grado de tratamiento del dolor se mide por el uso de una
escala de puntuación verbal de 4 puntos (VRSpi) en la que 0 = ningún dolor, 1 = dolor leve, 2 = dolor moderado, 3 = dolor intenso.
64. El uso de acuerdo con la reivindicación 61, en la que el tratamiento se asocia con menos efectos secundarios en comparación con un tratamiento con una cantidad similar de opioide en una composición de liberación inmediata.
65. El uso de acuerdo con la reivindicación 64, en la que los efectos secundarios se seleccionan del grupo que consiste en sedación, náuseas, mareos, vértigo, estreñimiento crónico, retención de orina, prurito, transpiración, boca seca y dolor intercurrente.
66. Una composición farmacéutica, como la definida en cualquiera de las reivindicaciones 1 a 60, para utilizar como un medicamento.
67. Una composición farmacéutica, como la definida en cualquiera de las reivindicaciones 1 a 60, para tratar a un paciente que sufre de dolor sensible a un opioide.
68. Una composición farmacéutica de acuerdo con la reivindicación 67, donde la cantidad de opioide en una base diaria suficiente para tratar el dolor en el paciente es menor que la cantidad de opioide suficiente para tratar el dolor en una medida semejante mediante el uso de una composición de liberación inmediata.
69. Una composición farmacéutica de acuerdo con la reivindicación 68, en que el grado de tratamiento del dolor se mide por el uso de una escala de puntuación verbal de 4 puntos (VRSpi) en la que 0 = ningún dolor, 1 = dolor leve, 2 = dolor moderado, 3 = dolor intenso.
70. Una composición farmacéutica de acuerdo con la reivindicación 69, en la que el tratamiento se asocia con menos efectos secundarios en comparación con un tratamiento con una cantidad similar de opioide en una composición de liberación inmediata.
71. Una composición farmacéutica de acuerdo con la reivindicación 70, en la que los efectos secundarios se seleccionan del grupo que consiste en sedación, náuseas, mareos, vértigo, estreñimiento crónico, retención de orina, prurito, transpiración, boca seca y dolor intercurrente.
Patentes similares o relacionadas:
Regímenes de dosificación de buprenorfina, del 1 de Julio de 2020, de Indivior UK Limited: Buprenorfina para su uso en un método para tratar el trastorno por uso de opioides en un ser humano que lo necesite, que comprende las etapas […]
 Forma de dosificación llena de líquido, disuasoria del abuso y de liberación inmediata, del 24 de Junio de 2020, de Pharmaceutical Manufacturing Research Services, Inc: Una cápsula disuasoria del abuso de liberación inmediata, que comprende:
(a) un principio activo susceptible de abuso;
(b) un primer polietilenglicol (PEG) […]
Forma de dosificación llena de líquido, disuasoria del abuso y de liberación inmediata, del 24 de Junio de 2020, de Pharmaceutical Manufacturing Research Services, Inc: Una cápsula disuasoria del abuso de liberación inmediata, que comprende:
(a) un principio activo susceptible de abuso;
(b) un primer polietilenglicol (PEG) […]
Gránulos de dispersión rápida, comprimidos de desintegración oral y métodos, del 3 de Junio de 2020, de Adare Pharmaceuticals, Inc: Microgránulos de dispersión rápida, farmacéuticamente aceptables, que tienen una mediana del tamaño de partícula en el rango de 100 μm a 300 […]
Pulverizador de buprenorfina sublingual, del 22 de Abril de 2020, de Fresh Cut Development, LLC: Una formulación de pulverización sublingual que comprende una cantidad efectiva de buprenorfina, o una sal farmacéuticamente aceptable de la misma, […]
Dispositivos de administración transdérmica resistentes al abuso y composiciones que comprenden un agonista de opioides y un derivado N-óxido no administrado por vía transdérmica de un antagonista de opioide para el tratamiento del dolor, del 22 de Abril de 2020, de EURO-CELTIQUE S.A.: Un dispositivo de administración transdérmica que comprende una composición farmacéutica, en el que dicha composición comprende un derivado […]
Formulación farmacéutica a base de ibuprofeno y codeína que tiene estabilidad mejorada, del 15 de Abril de 2020, de FARMASIERRA MANUFACTURING S.L: Formulación farmacéutica a base de ibuprofeno y codeínade estabilidad mejorada. La invención consiste en una nueva formulación farmacéuticaen forma […]
Parche transdérmico de oximorfona, del 8 de Abril de 2020, de Buzzz Pharmaceuticals Limited: Un parche transdérmico que tiene una capa que contiene el fármaco que comprende oximorfona o una sal farmacéuticamente aceptable de la misma, […]
Composiciones de buprenorfina y antagonistas del receptor de opioides mu, del 1 de Abril de 2020, de Alkermes Pharma Ireland Limited: Una composición que comprende buprenorfina y un antagonista del receptor opioide μ, en donde la composición está caracterizada por un Índice […]