Método para el cribado y/o evaluación de la eficacia de medicamentos para el tratamiento de enfermedades mitocondriales y síndrome MELAS.
La presente invención se refiere a un procedimiento para la identificación y evaluación de la eficacia de fármacos para el tratamiento de enfermedades que cursan con disfunción mitocondrial y/o síndrome de MELAS,
en un modelo de Saccharomices cerevisiae mutante A14G y en fibroblastos derivados de pacientes con síndrome MELAS y en cíbridos transmitocondriales MELAS.
Tipo: Patente de Invención. Resumen de patente/invención. Número de Solicitud: P201230936.
Solicitante: UNIVERSIDAD PABLO DE OLAVIDE.
Nacionalidad solicitante: España.
Inventor/es: SÁNCHEZ ALCÁZAR,José A, CORDERO MORALES,Mario, DE LA MATA,Mario, COTÁN MARÍN,David, OROPESA ÁVILA,Manuel, GARRIDO MARAVER,Juan.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C12Q1/04 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12Q PROCESOS DE MEDIDA, INVESTIGACION O ANALISIS EN LOS QUE INTERVIENEN ENZIMAS, ÁCIDOS NUCLEICOS O MICROORGANISMOS (ensayos inmunológicos G01N 33/53 ); COMPOSICIONES O PAPELES REACTIVOS PARA ESTE FIN; PROCESOS PARA PREPARAR ESTAS COMPOSICIONES; PROCESOS DE CONTROL SENSIBLES A LAS CONDICIONES DEL MEDIO EN LOS PROCESOS MICROBIOLOGICOS O ENZIMOLOGICOS. › C12Q 1/00 Procesos de medida, investigación o análisis en los que intervienen enzimas, ácidos nucleicos o microorganismos (aparatos de medida, investigación o análisis con medios de medida o detección de las condiciones del medio, p. ej. contadores de colonias, C12M 1/34 ); Composiciones para este fin; Procesos para preparar estas composiciones. › Determinación de la presencia o del tipo de microorganismo; Empleo de medios selectivos para la investigación o análisis de antibióticos o bactericidas; Composiciones para este fin que contienen un indicador químico.
- G01N33/532 FISICA. › G01 METROLOGIA; ENSAYOS. › G01N INVESTIGACION O ANALISIS DE MATERIALES POR DETERMINACION DE SUS PROPIEDADES QUIMICAS O FISICAS (procedimientos de medida, de investigación o de análisis diferentes de los ensayos inmunológicos, en los que intervienen enzimas o microorganismos C12M, C12Q). › G01N 33/00 Investigación o análisis de materiales por métodos específicos no cubiertos por los grupos G01N 1/00 - G01N 31/00. › Producción de compuestos inmunoquímicos marcados.
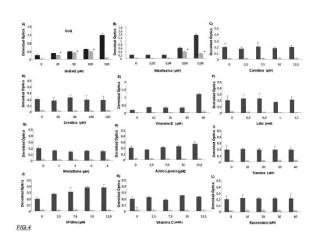
Fragmento de la descripción:
MÉTODO PARA EL CRIBADO Y/O EVALUACIÓN DE LA EFICACIA DE MEDICAMENTOS PARA EL TRATAMIENTO DE ENFERMEDADES MITOCONDRIALES Y SÍNDROME MELAS.
Campo de la invención La presente invención se encuadra en el campo general de la biomedicina y en particular se refiere a un método para el cribado y/o evaluación de la eficacia de un tratamiento para las enfermedades mitocondriales y síndrome MELAS.
Estado de la técnica Las enfermedades mitocondriales abarcan un amplio espectro de trastornos neurodegenerativos, crónicos y progresivos, con manifestaciones fenotípicas y grados de afección variables, como consecuencia de alteraciones en el metabolismo oxidativo mitocondrial [Zeviani M, Carelli V. Mitochondrial disorders. Curr Opin Neurol 2007;20:564-571]. A nivel celular, la patogénesis de estos desórdenes tiene su origen en un estado crónico de insuficiencia energética, debido a la incapacidad de las mitocondrias afectadas de generar suficiente ATP mediante el sistema OXPHOS (fosforilación oxidativa) . Como consecuencia, se produce una conversión de piruvato a lactato, que sistémicamente se manifiesta como una acidosis láctica crónica. A menudo, las citopatías mitocondriales presentan un patrón multisistémico, siendo los tejidos con una fuerte demanda energética como el cerebro y el músculo, los órganos que aparecen afectados con mayor frecuencia.
Los 37 genes del DNA mitocondrial (mtDNA) son imprescindibles para la fosforilación oxidativa. De éstos, 13 codifican subunidades de los complejos de la cadena respiratoria: siete subunidades del complejo I, una subunidad del complejo III, tres subunidades del complejo IV y dos subunidades del complejo V. Las mutaciones de estos genes causan diversas alteraciones mitocondriales y generalmente presentan herencia materna. Además, se requieren 22 tRNA y 2 RNA ribosomales (rRNA) para la síntesis proteica mitocondrial.
En la última década, los investigadores clínicos también se han interesado por las alteraciones de la mitocondria con herencia mendeliana. El DNA nuclear (nDNA) codifica un amplio número de genes necesarios para la fosforilación oxidativa, incluyendo 72 subunidades polipeptídicas, así como todos los factores requeridos para el ensamblaje correcto de la cadena respiratoria y la maquinaria necesaria para la integridad, replicación, reparación y expresión del mtDNA. Del mismo modo, las mutaciones en los factores que se requieren para la traducción de proteínas en la mitocondria, la importación de proteínas, y la fusión/fisión de mitocondrias también causan alteraciones mitocondriales [Debray FG, Lambert M, Mitchell GA. Disorders of mitochondrial function. Curr Opin Pediatr 2008;20:471-482].
Las enfermedades mitocondriales son clínicamente heterogéneas debido a la distribución desigual de las mutaciones en los distintos tejidos, al grado de heteroplasmia de los tejidos afectados, a la segregación mitótica y a la variabilidad de penetrancia y de efecto umbral de las distintas mutaciones. La prevalencia de las enfermedades mitocondriales es de aproximadamente 1:5000 entre la población a nivel mundial.
Actualmente no se dispone de tratamientos eficaces para la mayor parte de ellas [Stacpoole PW. Why are there no proven therapies for genetic mitochondrial diseases? Mitochondrion 2011], limitándose éstos a medidas paliativas, generales y farmacológicas. Por lo general, se trata de procesos degenerativos, pero pueden tener un curso crónico estacionario, en forma de manifestaciones crónicas recurrentes, pudiendo mostrar en ocasiones una mejoría espontánea hasta su recuperación.
El síndrome MELAS debe su nombre al acrónimo en inglés de Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes (encefalomiopatía mitocondrial, acidosis láctica y episodios semejantes a los accidentes cerebrovasculares) . Fue descrito por primera vez por Pavlakis et al., en 1984. Los pacientes presentan unas manifestaciones clínicas que incluyen la tríada de síntomas que dan nombre a la enfermedad. Los accidentes cerebrovasculares afectan principalmente la región parieto-occipital del cerebro lo que conduce a defectos en el campo visual. Las convulsiones son comunes en estos pacientes asociadas a episodios de ictus o como un fenómeno aislado. Otras manifestaciones incluyen migrañas intermitentes, vómitos, depresión, ataxia, trastornos cognitivos, baja estatura, sordera, intolerancia al ejercicio, cardiomiopatía y diabetes mellitas [Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelikeepisodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Annals of the New York Academy of Sciences 2008;1142:133-158].
La edad de aparición de esta enfermedad se ha descrito inicialmente dentro del rango entre los 2 y los 60 años, aunque casi el 70% de los pacientes presentan los síntomas iniciales entre los 2 y los 20 años. La progresión de la enfermedad es frecuentemente dramática y los pacientes experimentan un progresivo deterioro neurológico y neuromuscular que resulta en demencia, severa invalidez y muerte repentina, a menudo antes de los 20 años. La supervivencia media después del diagnóstico es de 6, 5 años. Dada la alta morbilidad y mortalidad del síndrome MELAS es necesaria la búsqueda de nuevos tratamientos capaces mejorar el pronóstico de la enfermedad.
El síndrome MELAS se trata de un desorden poligénico, asociado con al menos 29 mutaciones puntuales específicas en el mtDNA. La mutación más común relacionada con este síndrome, que supone un 80% de los casos, es la transición de una adenina a una guanina en la posición 3243 del genoma mitocondrial (A3243G) , en el gen que codifica para el ARNtLeu (UUR) , con una prevalencia de 0, 06% de la población general [Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Annals of the New York Academy of Sciences 2008;1142:133-158]. Por otra parte, se han asociado a esta enfermedad al menos otras siete mutaciones puntuales en dicho gen, mutaciones en otros genes de ARNt (His, Lys, Gln y Glu) y en genes que codifican proteínas (MT-ND1, MT-CO3, MTND4, MT-ND5, MT-ND6 y MT-CYB) [Wong LJ. Pathogenic mitochondrial DNA mutations in protein-coding genes. Muscle & nerve 2007;36:279-293]. Muchas de estas mutaciones, particularmente las que afectan a subunidades proteicas, están implicadas en otros síndromes mitocondriales (Neuropatía Óptica Hereditaria de Leber (LHON) , Enfermedad de Leigh, Epilepsia Mioclónica asociada a Fibras Rojo-Rasgadas (MERRF) ) .
La mutación A3243G dificulta la modificación de la base U de balanceo, entorpeciendo la traducción de los codones UUA y UUG, lo que resulta en una incorporación alterada de los aminoácidos a las proteínas sintetizadas en la mitocondria [Kirino Y, Yasukawa T, Ohta S, Akira S, Ishihara K, Watanabe K et al. Codonspecific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proceedings of the National Academy of Sciences of the United States of America 2004;101:15070-15075]. Otros factores propuestos que también influyen en la síntesis alterada de proteínas mitocondriales son: trastornos en el procesamiento de los mRNA, cinética de aminoacilación incorrecta de los tRNALeu (UUR) o conjugación incorrecta de los aminoácidos a tRNALeu (UUR) .
En esta enfermedad existe una deficiencia generalizada en la síntesis de proteínas mitocondriales, una disminución de la actividad de las enzimas de la cadena respiratoria mitocondrial y graves defectos respiratorios. Al menos el 42% de los pacientes con MELAS muestran una disminución de la actividad del complejo I, seguido de un 29% con disminución del complejo III y un 23% en el complejo IV [Santa KM. Treatment options for mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. Pharmacotherapy 2010;30:1179- 1196.]
En presencia de una cadena respiratoria disfuncional las mitocondrias no son capaces de producir suficientes cantidades de ATP. Esto conduce a un estado crónico de deficiencia energética, debido a un desequilibrio entre los requerimientos energéticos y la energía disponible. Finalmente, este desequilibrio energético causa daño celular y tisular. En general, la mutación A3243G provoca una mayor tasa glicolítica, aumento de la producción de lactato, oxidación reducida de la glucosa, una respuesta alterada a NADH, baja f4m, una producción disminuida de ATP, aumento de ROS y una homeostasis del calcio intracelular alterada, disminución en la secreción de insulina, envejecimiento prematuro y una desregulación del metabolismo de los aminoácidos y la síntesis...
Reivindicaciones:
1. Procedimiento para la identificación y evaluación de la eficacia de fármacos para el tratamiento de enfermedades que cursan con disfunción mitocondrial y/o síndrome de MELAS caracterizado porque comprende los siguientes pasos:
a) cribado de fármacos en un modelo de Saccharomices cerevisiae mutante A14G, mediante la exposición de dichas levaduras a al menos un fármaco y determinar si dicho fármaco produce crecimiento celular de las levaduras mutantes A14G,
b) evaluación de la eficacia del fármaco que produce crecimiento celular de las levaduras mutantes A14G del paso a) en modelos celulares derivados de pacientes con síndrome MELAS y determinar si el fármaco es eficaz
mediante la capacidad que tiene dicho fármaco para restaurar las alteraciones fisiopatológicas de dichos modelos celulares,
2. Procedimiento según la reivindicación 1, caracterizado porque los modelos celulares derivados de pacientes con síndrome MELAS del paso b) , son fibroblastos derivados de pacientes con síndrome MELAS y/o en cíbridos transmitocondriales MELAS.
3. Procedimiento según cualquiera de las reivindicaciones anteriores, caracterizado porque las alteraciones fisiopatológicas restauradas en los modelos celulares del paso b) son un aumento de la proliferación celular, un aumento de los niveles de ATP, una disminución de ROS, una disminución de la actividad mitofágica, un aumento de la expresión de proteínas y/o aumento de la actividad mitocondrial.
4. Procedimiento según cualquiera de las reivindicaciones anteriores, caracterizado porque el crecimiento celular 20 de las levaduras mutantes A14G del paso a) se realiza mediante densidad óptica.
Patentes similares o relacionadas:
Método, sistema y producto del programa informático para determinar la presencia de microorganismos e identificar dichos microorganismos, del 29 de Julio de 2020, de BIOMERIEUX: Un método para determinar la presencia de al menos un microorganismo determinado en una placa de Petri que comprende una o más colonias de microorganismos y un medio de […]
Preparación de células biológicas sobre soportes de muestras para espectrometría de masas para una ionización por desorción, del 13 de Mayo de 2020, de Bruker Daltonik GmbH: Procedimiento para la preparación de proteínas a partir de muestras de células biológicas no purificadas sobre un soporte de muestras para una determinación mediante […]
Disposición de detector para botellas de cultivo de sangre con sensores colorimétricos, del 13 de Mayo de 2020, de BIOMERIEUX, INC.: Un dispositivo de detección que comprende: una botella de cultivo de sangre que incorpora un sensor colorimétrico sujeto a cambio de color debido a un cambio […]
Método mejorado para la determinación de microorganismos, del 6 de Mayo de 2020, de STORA ENSO OYJ: Un método para determinar el contenido de microorganismos en un material que comprende celulosa en la industria de la pulpa y el papel que comprende las etapas de: […]
Procedimiento para detectar bacterias coliformes contenidas en la leche, del 15 de Abril de 2020, de ASAHI KASEI KABUSHIKI KAISHA: Procedimiento para lisar bacterias coliformes contenidas en la leche, que comprende la etapa de mezclar un agente de lisis que contiene lisozima, un surfactante aniónico […]
Protocolos de rastreo de lotes múltiples de composiciones modulares para la detección de patógenos, contaminantes microbianos y/o constituyentes, del 25 de Marzo de 2020, de INSTITUTE FOR ENVIRONMENTAL HEALTH, INC: Un procedimiento de muestreo, ensayo y validación de lotes de ensayo, que comprende: a) recoger una pluralidad de porciones de cada una de una pluralidad de lotes de ensayo, […]
Composiciones y procedimientos para identificar especies de Ehrlichia, del 18 de Marzo de 2020, de Abaxis, Inc: Un procedimiento de identificación de las especies de Ehrlichia que infectan a un sujeto, si están presentes, comprendiendo el procedimiento: poner en contacto una […]
Métodos y grupos, del 11 de Marzo de 2020, de Genome Research Limited: Un método para la identificación de cepas aisladas bacterianas adecuadas para su uso en bacterioterapia, comprendiendo el método: (i) preparar una suspensión […]