COMPUESTOS PEPTIDICOS ANTIBACTERIANOS.
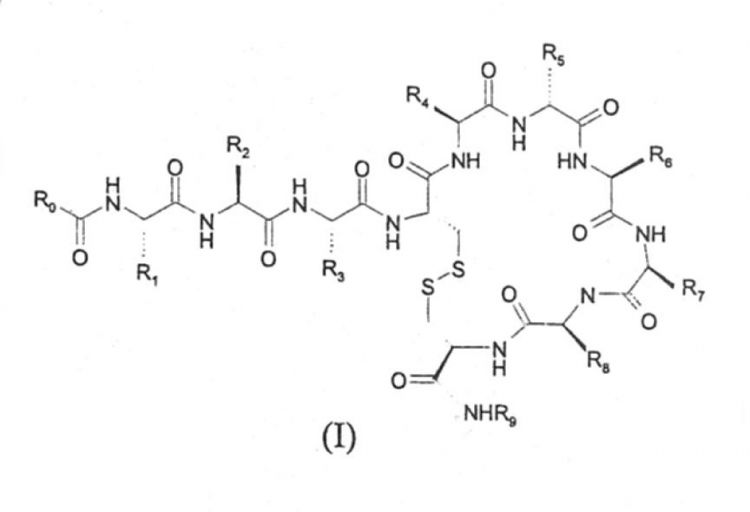
Compuestos peptídicos antibacterianos.Los compuestos de fórmula (I),
o retroenantiómeros de los mismos no acilados en su extremo N-terminal y con la configuración del aminoácido con la cadena lateral R{sub,2} no invertida cuando R{sub,2} es -CH(CH{sub,3})(OH), donde: R{sub,0} es CH{sub,3}-(CH{sub,2}){sub,m}-, CH{sub,3}-O-(CH{sub,2}CH{sub,2}O)2CH{sub,2}- o fenil-(CH{sub,2}){sub,x}-; m es un entero entre 7 y 10; x es un entero entre 1 y 3; R{sub,1}, R{sub,3}, R{sub,4}, R{sub,7} y R{sub,8} se seleccionan independientemente de la fórmula GF(CH{sub,2}){sub,n}- donde n es un entero entre 1 y 4; y GF es -NH{sub,2}, -NH-C(=NH)-NH{sub,2} o 4 imidazolilo; R{sub,2} es -CH(CH{sub,3})(OH), -CH(CH{sub,3}){sub,2}, -CH{sub,2}NH{sub,2} o -CH{sub,2}OH; R{sub,5} y R{sub,6} son (C{sub,1}-C{sub,4})-alquilo lineal o ramificado, -(CH{sub,2})-R{sub,10}, -CH{sub,2}-CH{sub,2}-S-CH{sub,3} o -CH{sub,2} CH{sub,2} S(=O)-CH{sub,3}; R{sub,9} es H o Gly-espermida; y R{sub,10} es fenilo, 3-indolilo, 4 imidazolilo, 4-hidroxifenilo, alfa o beta-naftilo o 2-, 3- ó 4-piridilo, se ha encontrado que son útiles en el tratamiento de infecciones bacterianas
Tipo: Patente de Invención. Resumen de patente/invención. Número de Solicitud: P200802626.
Solicitante: UNIVERSIDAD DE BARCELONA.
Nacionalidad solicitante: España.
Provincia: BARCELONA.
Inventor/es: RABANAL ANGLADA,FRANCESC, CAJAL VISA,YOLANDA, RODRIGUEZ NUÑEZ,MONTSERRAT, GARCIA SUBIRATS,MARIA.
Fecha de Solicitud: 10 de Septiembre de 2008.
Fecha de Publicación: .
Fecha de Concesión: 23 de Noviembre de 2010.
Clasificación Internacional de Patentes:
- A61K38/12 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 38/00 Preparaciones medicinales que contienen péptidos (péptidos que contienen ciclos beta-lactama A61K 31/00; dipéptidos cíclicos que no tienen en su molécula ningún otro enlace peptídico más que los que forman su ciclo, p. ej. piperazina 2,5-dionas, A61K 31/00; péptidos basados en la ergolina A61K 31/48; que contienen compuestos macromoleculares que tienen unidades aminoácido repartidas estadísticamente A61K 31/74; preparaciones medicinales que contienen antígenos o anticuerpos A61K 39/00; preparaciones medicinales caracterizadas por los ingredientes no activos, p. ej. péptidos como soportes de fármacos, A61K 47/00). › Péptidos cíclicos.
- C07K7/62 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07K PEPTIDOS (péptidos que contienen β -anillos lactamas C07D; ipéptidos cíclicos que no tienen en su molécula ningún otro enlace peptídico más que los que forman su ciclo, p. ej. piperazina diones-2,5, C07D; alcaloides del cornezuelo del centeno de tipo péptido cíclico C07D 519/02; proteínas monocelulares, enzimas C12N; procedimientos de obtención de péptidos por ingeniería genética C12N 15/00). › C07K 7/00 Péptidos con 5 a 20 aminoácidos en una secuencia totalmente determinada; Sus derivados. › Polimixinas; Péptidos semejantes.
Clasificación PCT:
- A61K38/12 A61K 38/00 […] › Péptidos cíclicos.
- A61P31/04 A61 […] › A61P ACTIVIDAD TERAPEUTICA ESPECIFICA DE COMPUESTOS QUIMICOS O DE PREPARACIONES MEDICINALES. › A61P 31/00 Antiinfecciosos, es decir antibióticos, antisépticos, quimioterápicos. › Agentes antibacterianos.
- C07K7/62 C07K 7/00 […] › Polimixinas; Péptidos semejantes.
Fragmento de la descripción:
Compuestos peptídicos antibacterianos.
La presente invención está relacionada con compuestos que son activos contra bacterias Gram-positivas y Gram-negativas, su procedimiento de preparación, composiciones farmacéuticas que los contienen, y su uso en el tratamiento de infecciones bacterianas.
Estado de la técnica
El hecho de que ciertos microorganismos patógenos se hayan convertido en resistentes a las terapias antibióticas es un problema grave en la salud pública. Parte de este problema radica en el hecho de que ciertas bacterias y otros microorganismos infecciosos son extraordinariamente capaces de desarrollar resistencia a los antibióticos. Otra causa principal se debe al uso deficiente de los antibióticos en medicina, veterinaria y agricultura.
Existe una preocupación a nivel mundial por la creciente prevalencia de infecciones causadas por bacterias multiresistentes, como por ejemplo Staphylococcus aureus resistente a metilicina, los Enterococcus resistentes a vancomincina y ciertas bacterias Gram-negativas como Pseudomonas aeruginosa, Acinetobacter baumanii y Klebsiella pneumoniae. Dichas infecciones son muy difíciles de controlar y una causa constante de enfermedad y mortandad. Los antibióticos convencionales actúan habitualmente sobre una o mas proteínas o receptores diana y la resistencia genética aparece a una frecuencia que depende de muchos factores, tales como el número de dichas proteínas o receptores diana.
La continua aparición de cepas bacterianas resistentes a los antibióticos convencionales está llevando a enormes esfuerzos dirigidos al desarrollo de nuevos fármacos que actúen en la membrana bacteriana, como los péptidos antimicrobianos ("anti-microbial peptides", AMP). Los péptidos antimicrobianos ofrecen una nueva clase de agentes terapéuticos a los cuales las bacterias no son capaces de desarrollar resistencia genética, puesto que actúan principalmente sobre el componente lipídico de las membranas celulares. Entre dichos compuestos, la polimixina B (PxB), que se halla aprobada para su uso clínico, esta adquiriendo una nueva relevancia terapéutica y está empezando a ser considerado como un representante de la clase de antibióticos activos contra bacterias multiresistentes.
Las polimixinas, en particular la polimixina B, constituyen una familia de antibióticos descubierta en 1947 con una elevada actividad contra bacterias Gram-negativas. La polimixina B es un lipopéptido antibiótico aislado de Bacillus polymyxa. Su estructura básica consiste en un ciclo peptídico policatiónico del cual pende un tripéptido unido a una cadena de ácido graso. La polimixina B ha resurgido en la práctica médica durante los últimos años y su uso continuará en aumento debido al escaso desarrollo de nuevos antibióticos por parte de la compañías farmacéuticas y a la creciente prevalencia mundial de infecciones nosocomiales causadas por bacterias Gram-negativas multiresistentes ("multidrug resistant", MDR). La polimixina B y otros miembros de la familia de las polimixinas son fármacos que se utilizan como último remedio para tratar infecciones causadas por bacterias multiresistentes y algunas veces son el único antibiótico activo disponible. Además, la resistencia a polimixina es rara y en general, adaptativa y, por tanto, reversible. La polimixina B es también capaz de inhibir la actividad biológica del lipopolisacárido (LPS) bacteriano por medio de la unión de alta afinidad al lípido A, siendo así el agente de elección para el tratamiento del shock séptico inducido por LPS. Desgraciadamente, la polimixina B no presenta actividad contra bacterias Gram-positivas o anaeróbicas. Además, las polimixinas son de uso limitado debido a que presentan cierta nefrotoxicidad y neurotoxicidad.
Por todo ello, existe todavía la necesidad de encontrar nuevos agentes antibacterianos activos no sólo contra bacterias Gram-negativas sino también contra bacterias Gram-positivas.
Explicación de la invención
Los inventores han encontrado algunos compuestos peptídicos con actividad antibiótica que actúan sobre el componente lipídico de las membranas bacterianas y que son activos tanto contra bacterias Gram-positivas como contra Gram-negativas. Estos compuestos se basan en la estructura de la polimixina natural. Sin embargo y a diferencia de las polimixinas, dichos compuestos son activos no sólo contra bacterias Gram-negativas sino también en Gram-positivas en el rango micromolar. Esto es ventajosos puesto que pueden actuar en respuesta a infecciones causadas por ambos tipos de bacteria.
Así, un aspecto de la presente invención es el proporcionar compuestos de fórmula (I),

o retroenantiómeros de los mismos de fórmula (I'),

donde
R0 es un radical seleccionado entre CH3-(CH2)m-, CH3-O-(CH2CH2O)2CH2-, y

m es un entero entre 7 y 10; x es un entero entre 1 y 3; R1, R3, R4, R7, y R8 son radicales seleccionados independientemente que responden a la fórmula: GF-(CH2)n-; n es un entero entre 1 y 4; GF es un radical seleccionado entre -NH2(amino), -NH-C(=NH)-NH2(guanidino) y 4-imidazolilo; R2 es un radical seleccionado entre -CH(CH3)(OH), -CH(CH3)2, -CH2NH2 y -CH2OH; R5 y R6 son radicales seleccionados entre: -(C1-C4)-alquilo lineal o ramificado; -(CH2)-R10, -CH2-CH2-S-CH3, y -CH2-CH2-S(=O)-CH3, R9 es H o Gly-NHCH2CH2CH2NHCH2CH2CH2CH2NHCH2CH2CH2 NH2(Gly-espermida); y R10 es un radical seleccionado entre fenilo, 3-indolilo, 4-imidazolilo, 4-hidroxifenilo, naftilo y piridilo; con la condición de que el compuesto de fórmula (I) no es uno de los compuestos de la siguiente lista, que se han descrito previamente, pero no su aplicación como agentes antibacterianos contra bacterias Gram-positivas:
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Dab-Dab-Cys], (sp-B);
nonanoil-Dap-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Dap-Dab-Cys], (sP-D);
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Met-Dab-Dab-Cys], (sp-Met);
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Met(O)-Dab-Dab-Cys]; (sp-Met(O)); o
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DTrp-Leu-Dab-Dab-Cys], (sp-Bw).
Por el término "ciclo(S-S)" se entiende un macrociclo formado por un puente disulfuro entre las dos cisteínas.
Un retroenantiómero de un péptido es un análogo peptídico que consiste en la secuencia de aminoácidos reversa respecto al péptido original con inversión simultánea de la configuración de los estereocentros, en particular los del carbono alfa de cada aminoácido. Es conocido que los retroenantiómeros de ciertos péptidos y depsipéptidos cíclicos mantienen la actividad biológica a pesar de que la dirección del enlace amida (definido en el sentido del átomo de carbono del carbonilo al átomo de nitrógeno) que une los residuos se haya invertido (cf. M. Goodman et al.,"On the concept of linear modified retro-peptide structures", Acc. Chem. Res. 1979 vol. 12, pp. 1-7). La actividad biológica se atribuye a la orientación tridimensional de las cadenas laterales y no al esqueleto peptídico.
El compuesto retroenantiomérico (I') es un péptido análogo que consiste en la secuencia de aminoácidos reversa del compuesto (I) con inversión simultánea de la configuración de todos los centros quirales excepto el del aminoácido con la cadena lateral R2 cuando dicho R2 es treonina, y que no necesariamente se halla acilado en su extremo N-terminal.
En una realización preferida, los compuestos...
Reivindicaciones:
1. Compuesto de fórmula (I),

o un retroenantiómero del mismo de fórmula (I'),

donde:
R0 es un radical seleccionado entre el grupo que consiste en:
CH3-(CH2)m-,
CH3-O-(CH2CH2O)2CH2-, y

m es un entero entre 7 y 10;
x es un entero entre 1 y 3;
R1, R3, R4, R7, y R8 son radicales seleccionados independientemente que tienen la fórmula siguiente:
donde
n es un entero entre 1 y 4; y GF es un radical seleccionado entre el grupo que consiste en -NH2, -NH-C(=NH)-NH2 y 4-imidazolilo;
R2 es un radical seleccionado entre el grupo que consiste en -CH(CH3)(OH), -CH(CH3)2, -CH2NH2 y -CH2OH;
R5 y R6 son radicales seleccionados independientemente entre el grupo que consiste en -(C1-C4)-alquilo lineal o ramificado, -(CH2)-R10, -CH2-CH2-S-CH3 y -CH2-CH2-S(=O)-CH3;
R9 es H ó Gly-NHCH2CH2CH2NHCH2CH2CH2CH2NHCH2CH2CH2NH2; y
R10 es un radical seleccionado entre el grupo que consiste en fenilo, 3-indolilo, 4-imidazolilo, 4-hidroxifenilo, α o β-naftilo y 2-, 3- o 4-piridilo;
con la condición de que el compuesto de fórmula (I) no es uno de los siguientes compuestos:
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Dab-Dab-Cys];
nonanoil-Dap-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Dap-Dab-Cys];
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Met-Dab-Dab-Cys];
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Met(0)-Dab-Dab-Cys]; o
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DTrp-Leu-Dab-Dab-Cys].
2. Compuesto según la reivindicación 1, donde R2 es -CH(CH3)(OH).
3. Compuesto según cualquiera de las reivindicaciones 1-2, que es el compuesto de fórmula (I).
4. Compuesto según la reivindicación 3, que se selecciona de entre los siguientes:
nonanoil-Arg-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Arg-Dab-Cys];
nonanoil-Arg-Thr-Arg-ciclo(S-S)[Cys-Dab-DPhe-Leu-Arg- Dab-Cys];
nonanoil-Arg(NO2)-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Arg(NO2)-Dab-Cys];
nonanoil-Arg-Thr-Arg-ciclo(S-S)[Cys-Arg-DPhe-Leu-Arg-Arg-Cys];
nonanoil-Dap[(C=NH)-NH2]-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Dap[(C=NH)-NH2]-Dab-Cys]; y
nonanoil-Arg-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Arg-Dab-Cys]-Gly-NHCH2CH2CH2NHCH2CH2CH2CH2 NHCH2CH2CH2NH2.
5. Compuesto según cualquiera de las reivindicaciones 1-2, que es el compuesto de fórmula (I').
6. Compuesto según la reivindicación 5, que se selecciona de entre los siguientes:
ciclo(S-S)[DCys-DDab-DArg-DLeu-Phe-DDab-DCys]DDab-DThr-DArg-octilamida; y
ciclo(S-S)[DCys-DDab-DDab-DLeu-Phe-DDab-DCys]DDab-DThr-DDab-octilamida.
7. Compuesto según cualquiera de las reivindicaciones 1-6, o uno de los siguientes compuestos:
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Dab-Dab-Cys];
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Dab-Leu-Dab-Cys];
nonanoil-Dap-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Dap-Dab-Cys];
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Met-Dab-Dab-Cys];
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Met(0)-Dab-Dab-Cys]; o
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DTrp-Leu-Dab-Dab-Cys];
para su uso como agente antibacteriano contra bacterias Gram-positivas.
8. Compuesto según la reivindicación 7, donde las bacterias Gram-positivas se seleccionan entre el grupo que consiste en Micobacterium phlei, Staphylococcus aureus y Bacillus cereus var. mycoides.
9. Compuesto según cualquiera de las reivindicaciones 1-6, para uso como agente antibacteriano contra bacterias Gram-negativas.
10. Compuesto según la reivindicación 9, donde las bacterias Gram-negativas se seleccionan entre el grupo que consiste en Salmonella tvphimurium, Pseudomonas aeruginosa y Escherichia coli.
11. Uso de un compuesto coma se ha definido en cualquiera de las reivindicaciones 1-6, o uno de los siguientes compuestos,
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Dab-Dab-Cys];
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Dab-Leu-Dab-Cys];
nonanoil-Dap-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Leu-Dap-Dab-Cys];
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Met-Dab-Dab-Cys];
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DPhe-Met(0)-Dab-Dab-Cys]; y
nonanoil-Dab-Thr-Dab-ciclo(S-S)[Cys-Dab-DTrp-Leu-Dab-Dab-Cys];
para la preparación de un medicamento para el tratamiento de una infección bacteriana causada por bacterias Gram-positivas en un mamífero, incluyendo un humano.
12. Uso según la reivindicación 11, donde las bacterias Gram-positivas se seleccionan entre el grupo que consiste en Micobacterium phlei, Staphylococcus aureus y Bacillus cereus var. mycoides.
13. Uso de un compuesto como se ha definido en cualquiera de las reivindicaciones 1-6, para la preparación de un medicamento para el tratamiento de una infección bacteriana causada por bacterias Gram-negativas en una mamífero, incluyendo un humano.
14. Uso según la reivindicación 13, donde las bacterias Gram-negativas se seleccionan entre el grupo que consiste en Salmonella typhimurium, Pseudomonas aeruginosa y Escherichia coli.
15. Composición farmacéutica que comprende una cantidad terapéuticamente efectiva de un compuesto como se ha definido en cualquiera de las reivindicaciones 1-6, junto a excipientes o portadores farmacéuticamente aceptables.
Patentes similares o relacionadas:
Inmunomoduladores, del 29 de Julio de 2020, de BRISTOL-MYERS SQUIBB COMPANY: Un compuesto de la fórmula (I) **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en donde: A se selecciona de **(Ver fórmula)** en donde: […]
Péptidos de unión beta amiloide y sus usos para el tratamiento y el diagnóstico de la demencia de Alzheimer, del 17 de Junio de 2020, de Priavoid GmbH: Péptido que contiene al menos una secuencia de aminoácidos que se une a especies beta amiloides y en el que la carga negativa del grupo carboxilo presente […]
Macrociclos peptidomiméticos, del 10 de Junio de 2020, de AILERON THERAPEUTICS, INC: Un macrociclo peptidomimético de la fórmula: **(Ver fórmula)** o una sal aceptable farmacéuticamente de este, en donde: cada uno de Xaa3, Xaa5, Xaa6, Xaa7, […]
Régimen de dosificación de IL-2 para tratar lupus eritematoso sistémico, del 27 de Mayo de 2020, de ASSISTANCE PUBLIQUE, HOPITAUX DE PARIS: Interleucina-2 (IL-2) para su uso en el tratamiento del lupus eritematoso sistémico en un sujeto humano, donde la IL-2 debe administrarse en una […]
Tratamiento de glucogenosis de tipo II, del 20 de Mayo de 2020, de DUKE UNIVERSITY: α-Glucosidasa ácida (GAA) humana recombinante producida en un cultivo de células de ovario de hámster chino para uso en un método de tratamiento de glucogenosis […]
Proceso para la purificación de daptomicina, del 6 de Mayo de 2020, de Cubist Pharmaceuticals LLC: Un método para purificar daptomicina que comprende: a) someter a la daptomicina a condiciones en las que una solución micelar de daptomicina se forma alterando el pH; y […]
Péptidos cíclicos de localización en NTCP y sus usos como inhibidores de entrada, del 6 de Mayo de 2020, de Ruprecht-Karls-Universität Heidelberg: Un péptido cíclico de la fórmula general Ia cyclo[(X)m-P-(Y)n] (Ia) en el que P es la secuencia de aminoácidos NPLGFXaaP (SEQ. ID NÚM.: […]
Lipopéptidos de alta pureza, micelas de lipopéptidos y procesos para preparar los mismos, del 6 de Mayo de 2020, de Cubist Pharmaceuticals LLC: Un método para purificar daptomicina a partir de moléculas o agregados de alto peso molecular, en donde la daptomicina se proporciona en forma micelar, dicho […]