Terapia anaplerótica de enfermedad de Huntington y otras enfermedades de poliglutamina.
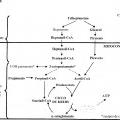
Precursor de propionil-CoA seleccionado del grupo que consiste en ácido graso de siete carbonos,
triheptanoína, heptanoato, 3-cetovalerato y 3-hidroxivalerato para su uso en un método para tratar y/o prevenir una enfermedad por poliglutamina.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/EP2007/063181.
Solicitante: INSERM (INSTITUT NATIONAL DE LA SANTE ET DE LA RECHERCHE MEDICALE).
Nacionalidad solicitante: Francia.
Dirección: 101, RUE DE TOLBIAC 75654 PARIS CEDEX 13 FRANCIA.
Inventor/es: DÜRR,ALEXANDRA, MOCHEL,FANNY.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A23L1/03
- A23L1/30
- A61K31/225 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › Acidos policarboxílicos.
PDF original: ES-2552843_T3.pdf




Patentes similares o relacionadas:
Método para la producción in situ de un emulsionante en un producto alimenticio, del 13 de Diciembre de 2017, de Dupont Nutrition Biosciences ApS: Un método para la producción in situ de un emulsionante en un producto alimenticio compuesto de 10-98% de agua, en donde el método comprende la etapa de añadir una lípido […]
Producción mejorada de lípidos que contienen ácidos grasos polienoicos por cultivos de alta densidad de microorganismos del orden Thraustochytriales en fermentadores, del 27 de Septiembre de 2017, de DSM IP ASSETS B.V.: Un procedimiento para producir lípidos microbianos eucariotas que comprende: (a) añadir una fuente de carbono no alcohol y una fuente de nutriente limitante a […]
Producción mejorada de lípidos que contienen ácidos grasos polienoicos por cultivos de alta densidad de microbios eucariotas en fermentadores, del 27 de Septiembre de 2017, de DSM IP ASSETS B.V.: Un método para enriquecer el contenido en ácidos grasos polienoicos de un microorganismo del orden Thraustochytriales que comprende fermentar […]
Métodos y composiciones para el control del peso y para mejorar el control glucémico, del 12 de Abril de 2017, de Gelesis LLC: Un hidrogel polimérico comestible que comprende carboximetilcelulosa reticulada con ácido cítrico para su uso en la potenciación del control glucémico […]
Uso de fosfatasa alcalina para la desintoxicación de LPS, del 22 de Marzo de 2017, de AM-PHARMA B.V.: El uso de fosfatasa alcalina para la fabricación de un medicamento para administrar a una capa mucosal del tracto gastrointestinal para la profilaxis o el […]
Material no viable derivado de probióticos, para la prevención y tratamiento de alergias, del 14 de Diciembre de 2016, de MJN U.S. Holdings, LLC: Un producto nutricional que comprende una composición que incluye una mezcla proteinácea, donde dicha composición es obtenible de un sobrenadante de cultivo en […]
Sistema de recubrimiento novedoso, del 30 de Noviembre de 2016, de DSM IP ASSETS B.V.: Un sistema de recubrimiento que comprende (i) al menos un compuesto lipídico y (ii) al menos una goma que tiene propiedades emulsionantes, […]
Procedimiento de preparación de un producto alimenticio enriquecido con probióticos y empobrecido en ácidos orgánicos, del 14 de Septiembre de 2016, de COMPAGNIE GERVAIS-DANONE: Procedimiento de preparación de un producto alimenticio acondicionado a base de frutas, caracterizado por que comprende las etapas siguientes: (a) empobrecimiento […]