Un método para identificar una muestra biológica para el análisis de la metilación.
Un método para identificar al menos una muestra biológica en el campo del análisis de la metilación,
que comprende las etapas (a) y (b) en el orden indicado:
(a) proporcionar un conjunto de muestras de al menos dos muestras biológicas, en donde al menos una muestra comprende el ADN genómico diferencialmente metilado al menos en una posición;
(b) aplicar al menos un identificador para cada muestra, en donde al menos un identificador aplicado no interfiera con el análisis posterior; y en donde al menos un identificador aplicado es un ácido nucleico que no forma una estructura secundaria estable y comprende al menos un sitio de unión de oligonúcleotidos libre de citosina, o libre de citosina y libre de guanina; que pone en contacto el ADN de cada muestra con el bisulfito;
(c) someter cada muestra a una reacción de detección o cuantificación específica para el(os) sitio(s) de unión de uno o más identificadores aplicados; y
(d) someter cada muestra al análisis de la metilación, en donde se detecta o cuantifica la metilación en donde la etapa (c) se lleva a cabo antes, simultáneamente con o después de la etapa (d).
Tipo: Patente Europea. Resumen de patente/invención. Número de Solicitud: E07090040.
Solicitante: EPIGENOMICS AG.
Nacionalidad solicitante: Alemania.
Dirección: Geneststrasse 5 10829 Berlin ALEMANIA.
Inventor/es: BERLIN, KURT, KLUTH, ANTJE, TETZNER,REIMO, DIETRICH,DIMO, WANDELL,Michael, SCHATZ,PHILIPP DR.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C12Q1/68 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12Q PROCESOS DE MEDIDA, INVESTIGACION O ANALISIS EN LOS QUE INTERVIENEN ENZIMAS, ÁCIDOS NUCLEICOS O MICROORGANISMOS (ensayos inmunológicos G01N 33/53 ); COMPOSICIONES O PAPELES REACTIVOS PARA ESTE FIN; PROCESOS PARA PREPARAR ESTAS COMPOSICIONES; PROCESOS DE CONTROL SENSIBLES A LAS CONDICIONES DEL MEDIO EN LOS PROCESOS MICROBIOLOGICOS O ENZIMOLOGICOS. › C12Q 1/00 Procesos de medida, investigación o análisis en los que intervienen enzimas, ácidos nucleicos o microorganismos (aparatos de medida, investigación o análisis con medios de medida o detección de las condiciones del medio, p. ej. contadores de colonias, C12M 1/34 ); Composiciones para este fin; Procesos para preparar estas composiciones. › en los que intervienen ácidos nucleicos.
PDF original: ES-2546848_T3.pdf


Fragmento de la descripción:
Un método para identificar una muestra biológica para el análisis de la metilación Campo de la invención La invención generalmente se relaciona con métodos nuevos y sustancialmente mejorados para identificar una muestra biológica para el análisis de la metilación que comprende un identificador particularmente su aplicación, uso y detección.
Antecedentes de aspectos de la invención Análisis de la metilación. Muchas enfermedades, particularmente las enfermedades de cáncer, se acompañan por una expresión modificada del gen. Ésta puede ser una mutación de los genes mismos, lo que conduce a una expresión de proteínas modificadas o a una inhibición o una sobre-expresión de las proteínas o enzimas. Una modulación de la expresión puede sin embargo ocurrir además por modificaciones epigenéticas particularmente la metilación de ADN. Tales modificaciones epigenéticas no afectan la actual secuencia codificante del ADN. Se ha encontrado que los procesos de metilación de ADN tienen implicaciones sustanciales para la salud, y parece ser claro que el conocimiento acerca de los procesos de metilación y modificaciones del metabolismo del metilo y metilación del ADN son esenciales para entender las enfermedades, para la profilaxis, diagnóstico y terapia de las enfermedades.
El control preciso de los genes, que representan sólo una parte pequeña del genoma completo de mamíferos, es una cuestión de la regulación bajo la consideración del hecho que la parte principal del ADN en el cromosoma es no codificante. La presencia de tal ADN tronco que contiene intrones, elementos repetitivos y elementos transponibles activamente en potencia, requiere mecanismos efectivos para su supresión duradera (silenciamiento) . Aparentemente, la metilación de citosina por metiltransferasas ADN dependientes S-adenosilmetionina (SAM) , que forma 5-metilcitosina, representa un mecanismo de ese tipo para la modificación de interacciones ADN-proteína. Los genes se pueden transcribir por promotores libres de metilación, aun cuando se metilan ampliamente las regiones transcritas o no transcritas adyacentes. Esto permite el uso y la regulación de promotores de genes funcionales, mientras que se suprime el ADN tronco que incluye los elementos transponibles. La metilación tiene lugar además por la supresión a largo plazo de genes enlazados a X y puede conducir ya sea a una reducción o un aumento del grado de transcripción, dependiendo de donde ocurre la metilación en la unidad de la transcripción.
Casi la metilación completa de ADN natural en mamíferos se restringe a secuencias palíndromo dinucleótido citosinaguanosina (CpG) que se controlan por metil transferasas de ADN. Los dinucleótidos CpG son aproximadamente 1 a 2 % de todos los dinucleótidos y se concentran en las llamadas islas de CpG. Una definición generalmente aceptada de islas de CpG significa que una región larga de ADN de 200 pb tiene un contenido de CpG de al menos 50 %, y que la relación del número de dinucleótidos CG observados y el número de los dinucleótidos CG esperados es mayor que 0.6 (Gardiner-Garden, M., Frommer, M. (1987) J. Mol. Biol. 196, 261-282. Típicamente, las islas de CpG tienen al menos 4 dinucleótidos CG en una secuencia que tiene una longitud de 100 pares de bases.
Si las islas de CpG están presentes en áreas del promotor, tienen frecuentemente una función reguladora para la expresión del respectivo gen. Si la isla de CpG es hipometilada, puede tener lugar la expresión. La hipermetilación frecuentemente conduce a la supresión de la expresión. En el estado normal, se hipometila un gene del supresor del tumor. Si tiene lugar una hipermetilación, esto conducirá a una supresión de la expresión del gen supresor del tumor, que es frecuentemente observado en tejidos de cáncer. En contraste a ella, los oncogenes se hipermetilan en el tejido saludable, mientras que frecuentemente se hipometilan en el tejido de cáncer.
Mediante la metilación de citosina, se previene regularmente la unión de proteínas que regulan la transcripción. Esto conduce a una modificación de la expresión génica. En cuanto al cáncer, por ejemplo la expresión de genes que regulan la división celular se afecta de ese modo, es decir por ejemplo la expresión de genes de apoptosis se regula negativamente, mientras que la expresión de oncogenes se regula positivamente. La hipermetilación del ADN tiene sin embargo además una influencia a largo plazo sobre la regulación. Mediante la metilación de la citosina, las proteínas de acetilación de histona pueden unirse por su dominio específico a ADN 5-metilcitosina. Esto tiene como consecuencia que las histonas son de-acetiladas, lo que conducirá a una compactación más apretada del ADN. De ese modo, las proteínas reguladoras no tienen más la posibilidad de unirse al ADN.
Por la razón de esto, la detección de la metilación del ADN es importante con respecto a diagnosticar una enfermedad, prognosticar una enfermedad, predecir una respuesta al tratamiento, diagnosticar una predisposición de una enfermedad, diagnosticar una progresión de una enfermedad, evaluar una enfermedad, estadificar una enfermedad, clasificar una enfermedad, caracterizar una enfermedad, o para identificar un nuevo marcador asociado con una enfermedad. Una visión general del método para el análisis de la metilación del ADN puede ser obtenida de Laird PW. "The power and the promise of DNA methylation markers" Nat Rev Cancer 2003 abril; 3 (4) :253-66. Muchos métodos para el análisis de la metilación se basan en el tratamiento del ADN genómico con el reactivo que diferencia entre las citosinas metiladas y no metiladas. En muchos casos, este reactivo es un reactivo de bisulfito que conduce a una conversión de citosinas no metiladas a uracilo o después de la amplificación a timina mientras que las citosinas metiladas permanecen sin cambios.
Necesidad pronunciada en la técnica. Al igual que muchos métodos modernos de flujos de trabajo de laboratorio para el análisis de la metilación se caracterizan en ese sentido un gran número de muestras que debe ser procesado. De ese modo es irrelevante, si se aplican métodos o flujos de trabajo para diagnosticar una enfermedad, pronosticar una enfermedad, predecir una respuesta al tratamiento, diagnosticar una predisposición de una enfermedad, diagnosticar una progresión de una enfermedad, evaluar una enfermedad, estadificar una enfermedad, clasificar una enfermedad, caracterizar una enfermedad, o identificar un nuevo marcador como metilación, ARN, o proteína que se asocia con una enfermedad. En cualquier caso, es importante que las muestras no se intercambien y ninguna muestra se contamine con otra muestra. Como técnica anterior relevante se considera lo siguiente:
De acuerdo con WO994385 las secuencias polimórficas endógenas se usan como identificador único para muestras biológicas. Estos identificadores enlazan la muestra a su fuente y a otra información relevante.
De acuerdo con la Patente de Estados Unidos 6, 153, 389 muestras forenses o médicas biológicas se marcan por la adición de ácidos nucleicos de secuencia conocida. Además utiliza iniciadores y su uso para la detección del ácido nucleico añadido en una reacción de amplificación resultando en una molécula de ácido nucleico de longitud específica.
Actualmente el solicitante no está consciente de cualquier técnica anterior, que aborda la cuestión de detectar el intercambio de muestra y/o contaminación cruzada de muestra para el análisis de la metilación. Los dos documentos citados anteriormente no enseñan un método para marcar una muestra biológica que resiste un tratamiento de bisulfito, tal como se usa muchas veces en el análisis de la metilación.
Resumen de aspectos de la invención Los aspectos de la presente descripción se relacionan con composiciones y métodos para identificar una muestra biológica en el campo del análisis de la metilación que comprende al menos un identificador, particularmente, su aplicación, uso y detección.
Aspectos particulares proporcionan composiciones y métodos para identificar una muestra biológica en el campo del análisis de la metilación, en donde se proporcionan muestras biológicas, uno o más identificadores se aplican a una muestra, el (los) identificador (es) aplicado (es) se detectan o cuantifican, y se analiza la metilación del ADN de cada muestra biológica.
Aspectos particulares proporcionan composiciones y métodos para identificar una muestra biológica en el campo del análisis de la metilación, en donde el ADN de la muestra biológica que incluye el identificador se pone en contacto con un reactivo o enzima que diferencia entre las posiciones de citosina metiladas o no metiladas.
Aspectos particulares... [Seguir leyendo]
Reivindicaciones:
1. Un método para identificar al menos una muestra biológica en el campo del análisis de la metilación, que comprende las etapas (a) y (b) en el orden indicado: 5
(a) proporcionar un conjunto de muestras de al menos dos muestras biológicas, en donde al menos una muestra comprende el ADN genómico diferencialmente metilado al menos en una posición;
(b) aplicar al menos un identificador para cada muestra, en donde al menos un identificador aplicado no interfiera con el análisis posterior; y en donde al menos un identificador aplicado es un ácido nucleico que no
forma una estructura secundaria estable y comprende al menos un sitio de unión de oligonúcleotidos libre de citosina, o libre de citosina y libre de guanina; que pone en contacto el ADN de cada muestra con el bisulfito;
(c) someter cada muestra a una reacción de detección o cuantificación específica para el (os) sitio (s) de unión de uno o más identificadores aplicados; y
(d) someter cada muestra al análisis de la metilación, en donde se detecta o cuantifica la metilación 15 en donde la etapa (c) se lleva a cabo antes, simultáneamente con o después de la etapa (d) .
2. Un método de la reivindicación 1, en donde la detección o cuantificación de uno o más identificadores aplicados para cada muestra y el análisis de la metilación de cada muestra se realizan simultáneamente.
genómico total derivado de un no vertebrado; una sección de ADN genómico o ADN genómico total derivado de un vertebrado; una repetición corta en tándem; una variante de un polimorfismo de deleción; una variante de un polimorfismo de nucleótido simple; una variante de un polimorfismo de longitud; un ácido nucleico artificial; un ácido nucleico circular; un ADN circular; un plásmido; un polinucleótido; un oligonucleótido; un PNA; un oligómero de PNA; un polímero de PNA; una metilación artificial; o combinaciones de estos.
4. Un método de la reivindicación 1, en donde diferentes identificadores se asignan a diferentes conjuntos de identificadores de acuerdo con sus respectivas propiedades biológicas, químicas o físicas.
5. Un método de la reivindicación 4, caracterizado además porque un representante de cada uno de al menos 35 dos conjuntos de identificadores está incluido en un plásmido.
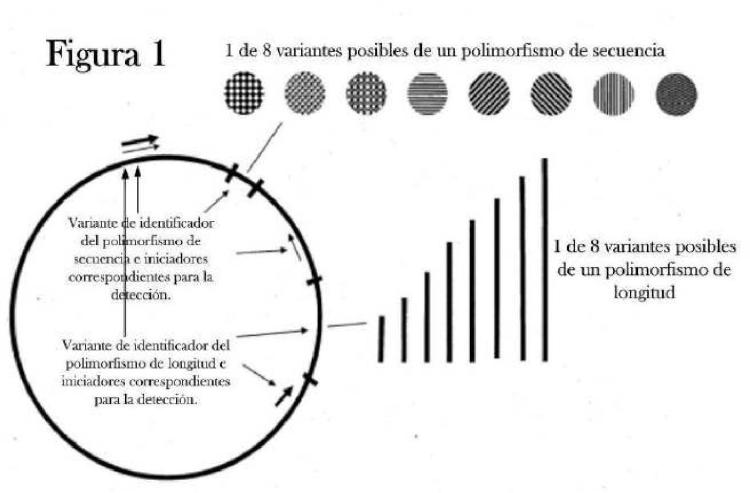
6. Un método de la reivindicación 5, en donde el primer conjunto de identificadores comprende un polimorfismo de secuencia y en donde el segundo conjunto de identificadores comprende un polimorfismo de longitud.
7. Un método de la reivindicación I, en donde el identificador comprende al menos un sitio de unión de oligonucleótidos que protege las posiciones de citosina convertidas por el bisulfito; es caracterizado además por una composición de base similar a la del ADN genómico analizado de la muestra proporcionada;
es una secuencia polimórfica de aproximadamente 5, aproximadamente 10, aproximadamente 15, aproximadamente 20, aproximadamente 25, aproximadamente 30, aproximadamente 35, aproximadamente 40, aproximadamente 50, aproximadamente 75, aproximadamente 100, o aproximadamente 200 nucleótidos; tiene un contenido de nucleótidos-citosina y nucleótidos-guanosina de aproximadamente 15 %, aproximadamente 20 %, aproximadamente 30 %, aproximadamente 40 %, aproximadamente 50 %,
aproximadamente 60 %, aproximadamente 70 %, o aproximadamente 80 %; o combinaciones de estos.
8. Un método de la reivindicación 1, en donde el identificador es caracterizado además por una composición de base similar a la del ADN genómico analizado de la muestra 55 proporcionada; es una secuencia polimórfica de aproximadamente 5, aproximadamente 10, aproximadamente 15, aproximadamente 20, aproximadamente 25, aproximadamente 30, aproximadamente 35, aproximadamente 40, aproximadamente 50, aproximadamente 75, aproximadamente 100, o aproximadamente 200 nucleótidos; tiene un contenido de nucleótidos-citosina y nucleótidos-guanosina de aproximadamente 15 %,
aproximadamente 20 %, aproximadamente 30 %, aproximadamente 40 %, aproximadamente 50 %, aproximadamente 60 %, aproximadamente 70 %, o aproximadamente 80 %; o combinaciones de estos.
9. Un método de la reivindicación 1, en donde el identificador es una variante de un polimorfismo de secuencia y, 65 además
comprende aproximadamente 1, aproximadamente 10, aproximadamente 20, aproximadamente 30, aproximadamente 40, aproximadamente 50, aproximadamente 60, aproximadamente 70, aproximadamente 80, aproximadamente 90, o aproximadamente 100 sitios de nucleótidos variables.
10. Un método de la reivindicación 1, en donde el identificador es una variante de un polimorfismo de secuencia y, además, comprende aproximadamente 5, aproximadamente 10, aproximadamente 15, aproximadamente 20, o aproximadamente 25 sitios nucleótidos variables; tiene un contenido de nucleótidos-citosina y nucleótidos-guanosina de aproximadamente 20 %, aproximadamente 30 %, aproximadamente 40 %, aproximadamente 50 %, aproximadamente 60 %, aproximadamente 70 %, o aproximadamente 80 %; o ambos.
11. Un método de la reivindicación 1, en donde el identificador es una variante de un polimorfismo de longitud y, además, tiene una diferencia de longitud de aproximadamente 10, aproximadamente 100, aproximadamente 200, aproximadamente 300, aproximadamente 400, aproximadamente 500, aproximadamente 600, aproximadamente 700, aproximadamente 800, aproximadamente 900, o aproximadamente 1000 nucleótidos en comparación con otros identificadores de polimorfismo de longitud de ácido nucleico usados; es de aproximadamente 10, aproximadamente 100, aproximadamente 500, aproximadamente 1.000, aproximadamente 1.500, aproximadamente 2.000, aproximadamente 2.500, aproximadamente 3.000, aproximadamente 3.500, aproximadamente 4.000, aproximadamente 4.500, aproximadamente 5.000, aproximadamente 5.500, aproximadamente 6.000, aproximadamente 6.500, aproximadamente 7.000, aproximadamente 7.500, aproximadamente 8.000, aproximadamente 8.500, aproximadamente 9.000, aproximadamente 9.500, o aproximadamente 10.000 nucleótidos de longitud; se deriva a partir del ADN nohumano; o combinaciones de estos.
12. Un método de la reivindicación 1, en donde el identificador es una variante de un polimorfismo de longitud y, además es de aproximadamente 5, aproximadamente 25, aproximadamente 50, aproximadamente 75, aproximadamente 100, aproximadamente 125, aproximadamente 150, aproximadamente 175, aproximadamente 200, aproximadamente 225, aproximadamente 250, aproximadamente 275, aproximadamente 300, aproximadamente 325, aproximadamente 350, aproximadamente 375, aproximadamente 400, aproximadamente 425, aproximadamente 450, aproximadamente 475, o aproximadamente 500 nucleótidos de longitud; se deriva a partir del ADN no-humano; tiene un contenido de nucleótidos-citosina y nucleótidos-guanosina de aproximadamente 20 %, aproximadamente 30 %, aproximadamente 40 %, aproximadamente 50 %, aproximadamente 60 %, aproximadamente 70 %, o aproximadamente 80 %; o combinaciones de estos.
13. Un método de la reivindicación 1, en donde el identificador comprende una etiqueta seleccionada del grupo que comprende colorante, colorante fluorescente, colorante quimioluminiscente, Cy5, Cy3, TAMRA, FAM, etiqueta, etiqueta de epítopo, péptido, polipéptido, proteína, sacárido, hormona, lípido, marcador de masa, partícula, partícula de oro, partícula de plata, partícula de platino, código embebido en parafina o combinaciones de estos.
14. Un método de la reivindicación 1, en donde la reacción de detección o cuantificación se lleva a cabo por uno o más medios seleccionados a partir del grupo que comprende: anticuerpo, análisis de inmunoelectrotransferencia Western, cromatografía, inmunoensayo, inmunoensayo ELISA, radioinmunoensayo, FPLC, HPLC, luz UV, luz, espectrómetro, MALDI-TOF, ácido nucleico, ADN, PNA, oligonucleótido, oligómero de PNA, método de amplificación, método de PCR, método de amplificación isotérmica, método de NASBA, método de LCR, método de amplificación específica de la metilación, método de MSP (PCR específica de la metilación) , método de MSP anidada, método de HeavyMethyl™, método de detección, método de detección específica de la metilación, método de secuenciación de bisulfito, detección por medio de matrices de ADN, detección por medio de micromatrices de oligonucleótidos, detección por medio de micromatrices de isla de CpG, detección por medio de enzimas de restricción, método de detección y amplificación simultánea específica de la metilación y, método de COBRA, PCR en tiempo real, método de PCR en tiempo real HeavyMethyl™, método de MSP Methylight™, método de Methylight™, método de Methylight™ AlgoTM, método de OM, método de Headloop Methylight™, método de HeavyMethyl™ Methylight™, método de HeavyMethyl™ ScorpionTM, método de MSP ScorpionTM, método de Headloop ScorpionTM, extensión del iniciador sensible a la metilación, y método Ms-SNuPE (extensión del iniciador de nucleótido simple sensible a la metilación) .
15. Un método de la reivindicación 1, en donde la reacción de detección o cuantificación comprende un ácido nucleico, ADN, PNA, oligonucleótido, o oligómero de PNA que
es al menos de aproximadamente 10, aproximadamente 15, aproximadamente 20, aproximadamente 25, aproximadamente 30, aproximadamente 35, aproximadamente 40, aproximadamente 50, aproximadamente 60, aproximadamente 70, aproximadamente 80, aproximadamente 90, aproximadamente 100, aproximadamente 150 o aproximadamente 200 nucleótidos de longitud, tiene un contenido de nucleótidos de citosina y nucleótidos de guanosina de aproximadamente 15 %, aproximadamente 20 %, aproximadamente 30 %, aproximadamente 40 %, aproximadamente 50 %, aproximadamente 60 %, aproximadamente 70 %, aproximadamente 80 %, o aproximadamente 85 %; tiene una temperatura de fusión de aproximadamente 37 °C, aproximadamente 45 °C, aproximadamente 50 °C, aproximadamente 55 °C, aproximadamente 60 °C, aproximadamente 65 °C, aproximadamente 70 °C, aproximadamente 75 °C, aproximadamente 80 °C, aproximadamente 85 °C, aproximadamente 90 °C, aproximadamente 95 °C, o aproximadamente 99 °C; o combinaciones de estos.
16. Un método de la reivindicación 1, en donde la reacción de detección o cuantificación comprende un oligonucleótido que es al menos de aproximadamente 5, aproximadamente 10, aproximadamente 15, aproximadamente 20, aproximadamente 25, aproximadamente 30, aproximadamente 35, aproximadamente 40, aproximadamente 50, aproximadamente 60, aproximadamente 70, aproximadamente 80, o aproximadamente 90 nucleótidos de longitud; tiene un contenido de nucleótidos-citosina y nucleótidos-guanosina de aproximadamente 5 %, de aproximadamente 10 %, aproximadamente 20 %, aproximadamente 30 %, aproximadamente 40 %, aproximadamente 50 %, aproximadamente 60 %, aproximadamente 70 %, aproximadamente 80 %, aproximadamente 90 % o aproximadamente 95 %; tiene una temperatura de fusión de aproximadamente 37 °C, aproximadamente 45 °C, aproximadamente 50 °C, aproximadamente 55 °C, aproximadamente 60 °C, aproximadamente 65 °C, aproximadamente 70 °C, aproximadamente 75 °C, aproximadamente 80 °C, aproximadamente 85 °C, aproximadamente 90 °C aproximadamente 95 °C, o aproximadamente 99 °C; o combinaciones de estos.
17. Un método de la reivindicación 1, en donde la reacción de detección o cuantificación comprende un oligonucleótido que es al menos de aproximadamente 16, aproximadamente 20, aproximadamente 25, aproximadamente 30, aproximadamente 35, o aproximadamente 40 nucleótidos de longitud; tiene un contenido de nucleótidos-citosina y nucleótidos-guanosina de aproximadamente 20 %, aproximadamente 30 %, aproximadamente 40 %, aproximadamente 50 %, aproximadamente 60 %, aproximadamente 70 %, o aproximadamente 80 %; y tiene una temperatura de fusión de aproximadamente 50 °C, aproximadamente 53 °C, aproximadamente 56 °C, aproximadamente 59 °C, o aproximadamente 62 °C.
18. Un método de la reivindicación 1, en donde la reacción de detección o cuantificación comprende un oligonucleótido que comprende una secuencia de iniciación específica del gen y una secuencia que se hibrida en una variante de un polimorfismo de secuencia.
19. Un método de la reivindicación 1, en donde la reacción de detección o cuantificación comprende un oligonucleótido que comprende dos dominios, en donde un dominio comprende una secuencia de iniciación específica del objetivo de aproximadamente 10, aproximadamente 15, aproximadamente 20, aproximadamente 25, aproximadamente 30, aproximadamente 35,
o aproximadamente 40 nucleótidos, tiene un contenido de nucleótidos-citosina y nucleótidos-guanosina de aproximadamente 20 %, aproximadamente 30 %, aproximadamente 40 %, aproximadamente 50 %, aproximadamente 60 %, aproximadamente 70 %, o aproximadamente 80 %, y tiene una temperatura de fusión de dominio de aproximadamente 50 °C, aproximadamente 52 °C, aproximadamente 54 °C, aproximadamente 56 °C, aproximadamente 58 °C, aproximadamente 60 °C, o aproximadamente 62 °C; y en la presente el otro dominio comprende una secuencia única de aproximadamente 5, aproximadamente 10, aproximadamente 15, aproximadamente 20, aproximadamente 25, aproximadamente 30, aproximadamente 35, aproximadamente 40, aproximadamente 45, o aproximadamente 50 nucleótidos, es libre de citosinas, guanina, o ambos, y tiene un contenido de nucleótidos-citosina y nucleótidos-guanosina de aproximadamente 20 %, aproximadamente 30 %, aproximadamente 40 %, aproximadamente 50 %, aproximadamente 60 %, aproximadamente 70 %, o aproximadamente 80 %.
20. Un método de la reivindicación 1, en donde el análisis de la metilación comprende al menos uno seleccionado del grupo que comprende la detección del estado de metilación, detección del nivel de la metilación, detección del patrón de la metilación, detección del nivel patrón de la metilación, método de amplificación, método de PCR, método de amplificación isotérmica, método de NASBA, método de LCR, método de amplificación específica de la metilación, método de MSP (PCR específica de la metilación) , método de MSP anidada, método de HeavyMethyl™, método de detección, método de detección específica de la metilación, método de secuenciación de bisulfito, detección por medio de matrices de ADN, detección por medio de micromatrices de
oligonuclótidos, detección por medio de micromatrices de isla de CpG, detección por medio de enzimas de restricción, método de detección y amplificación simultánea específica de la metilación y, método de COBRA, PCR en tiempo real, método de PCR en tiempo real HeavyMethyl™, método de MSP Methylight™, método de Methylight™, método de Methylight™ AlgoTM, método de OM, método de Headloop Methylight™, método de HeavyMethyl™ Methylight™, método de HeavyMethyl™ ScorpionTM, método de MSP ScorpionTM, método de Headloop ScorpionTM, extensión del iniciador sensible a la metilación, y método Ms-SNuPE (extensión del iniciador de nucleótido simple sensible a la metilación) .
21. Un método adecuado para la detección del intercambio de muestra, contaminación cruzada, o ambos en el campo de análisis de la metilación, que comprende las etapas del método de la reivindicación 1 y que comprende además las etapas de deducir la presencia de un intercambio de muestra o de una contaminación cruzada a partir de la presencia de al menos un identificador no aplicado a una muestra o la ausencia de un intercambio de muestra o una contaminación cruzada a partir de la ausencia de al menos un identificador no aplicado a una muestra.
22. Un método de la reivindicación 21, en donde la etapa de deducir la presencia o ausencia de un intercambio de la muestra, de una contaminación cruzada, o ambos comprende además deducir el grado de una contaminación cruzada para una única muestra a partir de la cantidad absoluta o relativa de al menos un identificador presente en dicha muestra única.
23. Un método adecuado para la identificación de una muestra en un conjunto de muestras mezclado en el campo de análisis de la metilación, que comprende las etapas del método de la reivindicación 1 y que comprende además las etapas para: identificar una muestra en el conjunto de muestras mezclado mediante la detección de al menos un identificador respectivo aplicado.
24. Un método adecuado para la detección de una inhibición de la amplificación en el campo de análisis de la metilación, que comprende las etapas del método de la reivindicación 1 y que comprende además las etapas de deducir una presencia, ausencia o inhibición parcial de la amplificación a partir de la presencia, ausencia o cantidad de el producto de una reacción de amplificación específica del identificador.
25. Un método adecuado para la normalización, calibración, o ambos en el campo de análisis de la metilación, que comprende las etapas del método de la reivindicación 1 y que comprende además las etapas para normalizar al menos una muestra, calibrar un procedimiento experimental, o ambos de acuerdo con uno o más identificadores detectados o cuantificados en comparación con la cantidad total añadida de uno o más identificadores.
26. Un método adecuado para la detección de una contaminación por arrastre en el campo de análisis de la metilación que comprende las etapas del método de la reivindicación 1 y que comprende además las etapas de deducir la presencia de una contaminación por arrastre de la muestra a partir de la presencia de al menos un identificador no aplicado en dicha muestra, o deducir la ausencia de una contaminación de la muestra de la ausencia de identificadores no aplicados en dicha muestra.
27. Un método adecuado para la evaluación del éxito de una etapa de hibridación en el campo de análisis de la metilación, en donde la reacción de detección o cuantificación comprende una etapa de hibridación, y el método comprende además las etapas del método de la reivindicación 1 y que comprende además las etapas de evaluar el éxito de una etapa de hibridación en donde (a) la presencia de una señal derivada de al menos un identificador aplicado indica la presencia de una etapa de hibridación exitosa, y en donde (b) la ausencia de señal derivada para al menos un identificador aplicado indica la presencia de una etapa de hibridación sin éxito.
28. Un método de las reivindicaciones 21, 23 a 27, que comprende además poner en contacto el ADN de cada muestra y al menos un identificador aplicado con un reactivo o enzima que diferencia entre una posición metilada o una no metilada.
29. Un método adecuado para el control de la precisión de un proceso o método, que comprende las etapas del método de la reivindicación 1, en donde
(a) se proporciona un conjunto de muestras de al menos 2, 3, 4, 100 200, 400 o 800 muestras biológicas;
(b) en donde los identificadores aplicados generan un patrón de identificación a través de la muestra; y en donde
(c) al menos un identificador se aplica a una muestra del conjunto; y el método que comprende además deducir la precisión de dicho proceso o método a partir de las señales de los identificadores detectados o cuantificados de las muestras.
30. Un método de la reivindicación 29, en donde sólo un único identificador se aplica a cada muestra del conjunto de la muestra.
31. Un método de la reivindicación 29, en donde deducir la precisión de dicho proceso o método a partir de las
señales de los identificadores detectados o cuantificados de las muestras, comprende determinar la presencia de un proceso o método libre de error, en donde dichas señales generan un patrón que se corresponde con el patrón de identificación generado inicialmente mediante la aplicación de los identificadores a las muestras; o determinar la ausencia de un proceso o método libre de error, en donde dichas señales generan un patrón que no se corresponde con el patrón de identificación generado inicialmente mediante la aplicación de los identificadores a las muestras.
32. Un método de la reivindicación 31, en donde el proceso o método es un proceso o método de gran productividad. 15
33. El uso de un método de acuerdo con una de las reivindicaciones anteriores para al menos uno seleccionado del grupo que comprende la detección del intercambio de muestra; detección de la contaminación cruzada; identificación de una muestra en un conjunto de muestras mezcladas; detección de inhibición de la amplificación; determinación de la velocidad de conversión del ADN; normalización de una muestra; calibración de una muestra; identificación de la contaminación por arrastre, control del éxito de una etapa de hibridación o combinaciones de estos.
Patentes similares o relacionadas:
Método para analizar ácido nucleico molde, método para analizar sustancia objetivo, kit de análisis para ácido nucleico molde o sustancia objetivo y analizador para ácido nucleico molde o sustancia objetivo, del 29 de Julio de 2020, de Kabushiki Kaisha DNAFORM: Un método para analizar un ácido nucleico molde, que comprende las etapas de: fraccionar una muestra que comprende un ácido nucleico molde […]
MÉTODOS PARA EL DIAGNÓSTICO DE ENFERMOS ATÓPICOS SENSIBLES A COMPONENTES ALERGÉNICOS DEL POLEN DE OLEA EUROPAEA (OLIVO), del 23 de Julio de 2020, de SERVICIO ANDALUZ DE SALUD: Biomarcadores y método para el diagnostico, estratificación, seguimiento y pronostico de la evolución de la enfermedad alérgica a polen del olivo, kit […]
Detección de interacciones proteína a proteína, del 15 de Julio de 2020, de THE GOVERNING COUNCIL OF THE UNIVERSITY OF TORONTO: Un método para medir cuantitativamente la fuerza y la afinidad de una interacción entre una primera proteína de membrana o parte de la misma y una […]
Secuenciación dirigida y filtrado de UID, del 15 de Julio de 2020, de F. HOFFMANN-LA ROCHE AG: Un procedimiento para generar una biblioteca de polinucleótidos que comprende: (a) generar una primera secuencia del complemento (CS) de un polinucleótido diana a partir […]
Métodos para la recopilación, estabilización y conservación de muestras, del 8 de Julio de 2020, de Drawbridge Health, Inc: Un método para estabilizar uno o más componentes biológicos de una muestra biológica de un sujeto, comprendiendo el método obtener un […]
Evento de maíz DP-004114-3 y métodos para la detección del mismo, del 1 de Julio de 2020, de PIONEER HI-BRED INTERNATIONAL, INC.: Un amplicón que consiste en la secuencia de ácido nucleico de la SEQ ID NO: 32 o el complemento de longitud completa del mismo.
Aislamiento de ácidos nucleicos, del 24 de Junio de 2020, de REVOLUGEN LIMITED: Un método de aislamiento de ácidos nucleicos que comprenden ADN de material biológico, comprendiendo el método las etapas que consisten en: (i) efectuar un lisado […]
Composiciones para modular la expresión de SOD-1, del 24 de Junio de 2020, de Biogen MA Inc: Un compuesto antisentido según la siguiente fórmula: mCes Aeo Ges Geo Aes Tds Ads mCds Ads Tds Tds Tds mCds Tds Ads mCeo Aes Geo mCes Te (secuencia […]