Oxidación en fase líquida optimizada.
Un proceso que comprende: oxidar un compuesto oxidable en una fase líquida de un medio de reacción multifásico contenido en uno o más reactores de oxidación inicial,
en el que dicho medio de reacción incluye un primer volumen continuo del 20 por ciento distinto que tiene una primera tasa de oxígeno-espacio-tiempo (oxígeno-STR) y un segundo volumen continuo del 20 por ciento distinto que tiene una segunda STR de oxígeno, en el que la relación de dicha primera STR de oxígeno con respecto a dicha segunda STR de oxígeno es al menos 1,5:1.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/US2005/030548.
Solicitante: Grupo Petrotemex, S.A. de C.V.
Nacionalidad solicitante: México.
Dirección: Ricardo Margain No. 444, Torre sur, Piso 16, Col. Valle de Campestre San Pedro Garza Garcia, Nuevo Leon 66265 MÉXICO.
Inventor/es: DE VREEDE, MARCEL, WONDERS,Alan,George, PARTIN,Lee Reynolds, STRASSER,Wayne Scott.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C07C51/21 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07C COMPUESTOS ACICLICOS O CARBOCICLICOS (compuestos macromoleculares C08; producción de compuestos orgánicos por electrolisiso electroforesis C25B 3/00, C25B 7/00). › C07C 51/00 Preparación de ácidos carboxílicos o sus sales, haluros o anhídridos. › con oxígeno molecular.
- C07C51/235 C07C 51/00 […] › de grupos —CHO o de grupos alcohol primario.
- C07C63/26 C07C […] › C07C 63/00 Compuestos que tienen grupos carboxilo unidos a los átomos de carbono de ciclos aromáticos de seis miembros. › Acido (1,4-)bencenodicarboxílico-1,4.
PDF original: ES-2552634_T3.pdf

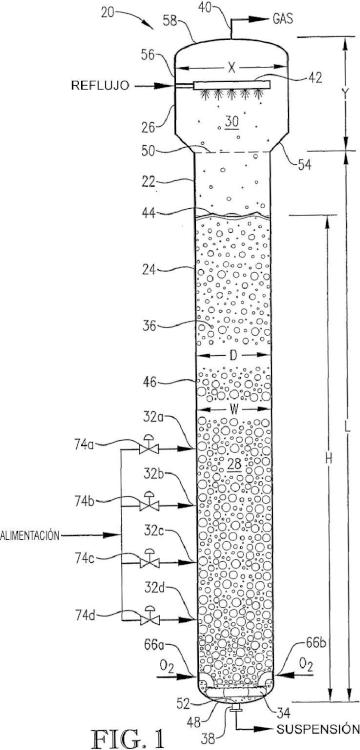
Patentes similares o relacionadas:
Método para producir ácido tereftálico de alta pureza, del 1 de Julio de 2020, de MITSUBISHI GAS CHEMICAL COMPANY, INC.: Método para producir ácido tereftálico de alta pureza, que comprende las siguientes etapas (a) a (c): la etapa (a); obtener un cristal […]
Método y aparato para el reciclaje de materiales poliméricos a través del proceso de despolimerización, del 11 de Marzo de 2020, de GR3N SAGL: Un método para el reciclaje de poliésteres y/o poliamidas a través del proceso de despolimerización, que comprende una reacción de despolimerización del material que se va […]
Sistema de oxidación con reactor secundario interno, del 22 de Enero de 2020, de Grupo Petrotemex, S.A. de C.V: Un reactor de columna de burbujeo que comprende: un recipiente de reacción externo ; y un recipiente de reacción […]
Recuperación de ácidos carboxílicos aromáticos y de catalizador de oxidación, del 1 de Enero de 2020, de INVISTA Textiles (U.K.) Limited: Un proceso que comprende: (a) producir una corriente de residuo de la fabricación de un ácido policarboxílico aromático por oxidación en fase líquida del precursor […]
Método de operación de filtración de tipo flujo transversal utilizando un filtro cerámico, del 11 de Diciembre de 2019, de MITSUBISHI GAS CHEMICAL COMPANY, INC.: Un método de operación de filtración para filtrar cristales finos contenidos en un licor madre de reacción de oxidación obtenido enfriando una suspensión de reacción […]
Procedimiento de enriquecimiento utilizando compuestos útiles en un procedimiento de poliéster, del 23 de Octubre de 2019, de Grupo Petrotemex, S.A. de C.V: Un procedimiento para producir una composición enriquecida , dicho proceso comprende las etapas (a), (f) y (g) y, opcionalmente, las etapas […]
Método de pretratamiento de una resina de quelato que tiene un anillo de piridina usada para recoger un catalizador en un procedimiento de producción de ácido tereftálico, del 24 de Abril de 2019, de MITSUBISHI GAS CHEMICAL COMPANY, INC.: Método de pretratamiento de una resina de quelato que contiene un anillo de piridina usada para recoger un catalizador de oxidación en fase líquida en un procedimiento […]
Sistema de producción de ácido policarboxílico que emplea digestión oxidativa con intercambio de aguas previo reducido o eliminado, del 27 de Marzo de 2019, de Grupo Petrotemex, S.A. de C.V: Un procedimiento para elaborar una composición de ácido tereftálico, comprendiendo dicho procedimiento: (a) introducir una alimentación de disolvente en una zona de oxidación […]