Análisis de tendencia en secuenciación.
Un método para reducir la tendencia de secuencia de una técnica de secuenciación que implica la unión con un adaptador,
comprendiendo el método:
(a) proporcionar un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 3' bloqueados (moléculas adaptadoras 3') y un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 5' bloqueados (moléculas adaptadoras 5'), donde las moléculas 3' y 5' comprenden uno o más nucleótidos degenerados;
(b) unir las moléculas adaptadoras 3' a los extremos 3' de las moléculas de ácido nucleico diana utilizando una ligasa;
(c) unir las moléculas adaptadoras 5' a los extremos 5' de las moléculas de ácido nucleico diana de la etapa (b) utilizando una ligasa; y
(d) determinar la secuencia de las moléculas de ácido nucleico diana que se obtienen en la etapa (c) utilizando un cebador capaz de hibridarse con el complemento de la molécula adaptadora 5' y un cebador capaz de hibridarse con la molécula adaptadora 3'.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/GB2012/051837.
Solicitante: UNIVERSITY OF EAST ANGLIA.
Nacionalidad solicitante: Reino Unido.
Dirección: Norwich Norfolk NR4 7TJ REINO UNIDO.
Inventor/es: SOREFAN,KARIM, DALMAY,TAMAS, MOULTON,VINCENT, PAIS,HELIO ERNESTO CORONEL MACHADO.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C12N5/10 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12N MICROORGANISMOS O ENZIMAS; COMPOSICIONES QUE LOS CONTIENEN; PROPAGACION, CULTIVO O CONSERVACION DE MICROORGANISMOS; TECNICAS DE MUTACION O DE INGENIERIA GENETICA; MEDIOS DE CULTIVO (medios para ensayos microbiológicos C12Q 1/00). › C12N 5/00 Células no diferenciadas humanas, animales o vegetales, p. ej. líneas celulares; Tejidos; Su cultivo o conservación; Medios de cultivo para este fin (reproducción de plantas por técnicas de cultivo de tejidos A01H 4/00). › Células modificadas por introducción de material genético extraño, p. ej. células transformadas por virus.
- C12Q1/68 C12 […] › C12Q PROCESOS DE MEDIDA, INVESTIGACION O ANALISIS EN LOS QUE INTERVIENEN ENZIMAS, ÁCIDOS NUCLEICOS O MICROORGANISMOS (ensayos inmunológicos G01N 33/53 ); COMPOSICIONES O PAPELES REACTIVOS PARA ESTE FIN; PROCESOS PARA PREPARAR ESTAS COMPOSICIONES; PROCESOS DE CONTROL SENSIBLES A LAS CONDICIONES DEL MEDIO EN LOS PROCESOS MICROBIOLOGICOS O ENZIMOLOGICOS. › C12Q 1/00 Procesos de medida, investigación o análisis en los que intervienen enzimas, ácidos nucleicos o microorganismos (aparatos de medida, investigación o análisis con medios de medida o detección de las condiciones del medio, p. ej. contadores de colonias, C12M 1/34 ); Composiciones para este fin; Procesos para preparar estas composiciones. › en los que intervienen ácidos nucleicos.
PDF original: ES-2550799_T3.pdf




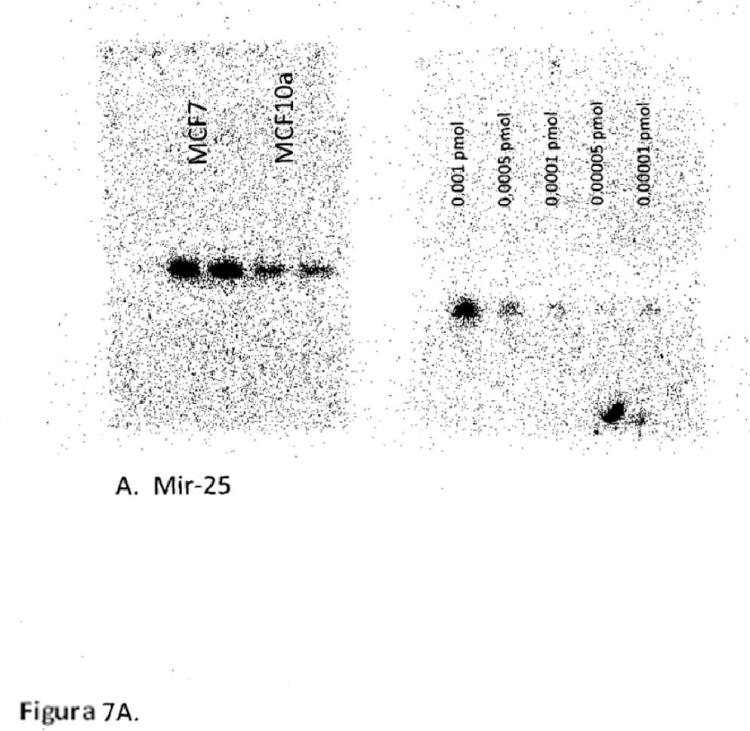
Fragmento de la descripción:
Análisis de tendencia en secuenciación Campo de la invención
La presente invención se refiere a métodos para determinar la tendencia de secuencia de una técnica de secuenciación. Además, la invención se refiere a métodos para reducir o aumentar la tendencia de secuencia durante la secuenciación de ácidos nucleicos por medio de técnicas que implican uniones a un adaptador. Específicamente el método se refiere al uso de una secuencia de ARN degenerado para analizar tendencias de secuencia cuando se generan bibliotecas de pequeños ARN, y al uso de adaptadores modificados para la clonación de pequeños ARN con secuencias degeneradas o específicas para reducir o aumentar la tendencia de la secuenciación, así como varias moléculas de ácidos nucleicos que se relacionan con estas o se derivan de las mismas.
Antecedentes de la invención
Muchos métodos biológicos moleculares necesitan una etapa de clonación que necesita el uso de una ADN o ARN ligasa para unir oligonucleótidos adaptadores u otras secuencias a una secuencia de nucleótido diana. La eficacia de esta unión depende de la secuencia del adaptador y la diana. Cuando se crea una biblioteca de secuencias a partir de ARN o ADN a menudo es importante unir todas las secuencias posibles y también que la biblioteca sea representativa de la abundancia relativa de la diana. Estas dos propiedades son importantes cuando se generan bibliotecas de alta calidad de pequeños ARN. Estas bibliotecas se pueden secuenciar utilizando la secuenciación tradicional de Sanger pero ahora se secuencian más comúnmente por secuenciación de alto rendimiento o técnicas de secuenciación de próxima generación (NGS).
La expresión genética en eucariotas está regulada en varias capas y uno de los mecanismos descubierto más recientemente implica moléculas de pequeños ARN no codificantes de 20-25 nucleótidos de longitud (sARN) (Fire, Xu et al. 1998; Voinnet 2002). Hay varias clases de sARN con diferentes rutas de biogénesis y modos de acción. La clase mejor caracterizada es la de los microARN (miARN) que se generan a partir de estructuras tallo-lazo y ARNm dianas in trans. La mayoría de los miARN se expresan específicamente en ciertos tejidos y en determinados estadios de desarrollo específicos y su acumulación a menudo cambia debido a señales externas y durante la enfermedad (MicroRNAs in Cáncer Translational Research, William.C.S.Cho, 2011, Springer). Varias bases de datos han recopilado la asociación de los miARN con la enfermedad que incluyen la base de datos de microARN enfermedad humana y la mir2enfermedad í http://www.mir2disease.org/ : http://202.38.126.151/ hmdd/ mirna/md/ 1 y se han lanzado recientemente nuevos productos para clasificar cánceres (o enfermedades) que se basan en sus perfiles de miARN, tales como Mirview (Rosetta Genomics, documento US 2010/0273172).
Por lo tanto, el perfil preciso del nivel de los miARN (y otras clases de sARN) es muy importante en la investigación básica y clínica. La identificación de un miARN interesante para estudios adicionales es un proceso empírico que a menudo se basa en su alta expresión y clara expresión diferencial. Estos criterios son más importantes cuando falta el contexto biológico del miARN tal como en animales en los que la predicción de la diana es pobre (Dalmay 2008). Además, el nivel de expresión se utiliza a menudo para discernir entre el miARN y el miARN estrella. El perfil de miARN preciso se complica por la naturaleza heterogénea de los miARN tal como las isoformas de secuencias y la longitud de los isomiros ya que se cree que estos tienen actividades diferenciales (Fernandez-Valverde, Taft et al. 2010; Guo y Lu 2010; Starega-Roslan, Kroletal. 2011).
La medición de los microARN con tecnologías de secuenciación tales como de alto rendimiento y Sanger o por PCR cualitativa (QPCR) necesita el uso de enzimas de modificación de ácidos nucleicos. Las ligasas, transcriptasas inversas y ADN polimerasas, son las enzimas más importantes que se utilizan en biología molecular. Para mejorar la actividad de estas enzimas se necesita entender completamente su función, lo que necesita un método para medir su actividad e identificar los determinantes que regulan su función.
El documento WO 2009/088933 describe métodos de mutagénesis que se aplican en particular en la generación de bibliotecas o matrices de proteínas mutantes o ácidos nucleicos que codifican tales proteínas. Los métodos de mutagénesis descritos implican el uso de cebadores que comprenden al menos un codón degenerado de 2 a 12 veces.
El informe de Christian ("RNA Captor: A Tool for RNA Characterization", PLOS ONE, vol. 6, n° 4, Abril 2011) describe un método de "mareaje" de ARN basado en el uso de T3 ARN ligasa 2 y adaptadores. Se describe un protocolo de RACE-PCR que se basa en cebadores específicos del adaptador y específicos genéticos.
Perelle et al. ("Comparison of PCR-ELISA and LightCycler real-time PCR assays for detecting Salmonella spp. in milk and meat samples", Molecular and Cellular Probes, vol. 18, n° 6, Diciembre 2004, páginas 409-420) expone los resultados de una comparación entre dos técnicas de PCR específicas para detectar Salmonella spp. Los oligonucleótidos cebadores y las sondas que se utilizan en este estudio se muestran en la Tabla 1.
Tian Geng et al. ("Sequencing bias: comparison of different protocols of microRNA library construction" BMC Biotechnology, vol. 10, n° 1, Septiembre 2010, página 64) describe una comparación de tres protocolos diferentes de preparación de una biblioteca de ARN. El protocolo SOLID utiliza adaptadores de doble cadena con un segmento protuberante N6 aleatorio degenerado.
Sumario de la invención
La presente invención proporciona un método para reducir la tendencia de secuencia de una técnica de secuenciación que implica la unión de un adaptador, comprendiendo el método:
(a) proporcionar un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 3 bloqueados (moléculas adaptadoras 3) y un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 5 bloqueados (moléculas adaptadoras 5) donde las moléculas adaptadoras 3 y 5 comprenden uno o más nucleótidos degenerados;
(b) unir las moléculas adaptadoras 3 a los extremos 3 de las moléculas de ácido nucleico diana utilizando una
ligasa;
(c) unir las moléculas adaptadoras 5 a los extremos 5 de las moléculas de ácido nucleico diana de la etapa (b)
utilizando una ligasa; y
(d) determinar la secuencia de las moléculas de ácido nucleico diana que se obtienen en la etapa (c) utilizando un cebador capaz de hibridarse con el complemento de la molécula adaptadora 5 y un cebador capaz de hibridarse con la molécula adaptadora 3.
En otro aspecto más, la presente invención proporciona un método para detectar preferentemente una molécula de ácido nucleico en una biblioteca de moléculas de ácido nucleico, comprendiendo el método:
(a) proporcionar un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 3 bloqueados (moléculas adaptadoras 3) y un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 5 bloqueados (moléculas adaptadoras 5) donde las moléculas adaptadoras 3 y 5 comprenden uno o más nucleótidos degenerados y que se pueden unir preferentemente a la molécula de ácido nucleico diana;
(b) unir las moléculas adaptadoras 3 a los extremos 3 de las moléculas de ácido nucleico diana utilizando una
ligasa;
(c) unir las moléculas adaptadoras 5 a los extremos 5 de las moléculas de ácido nucleico diana de la etapa (b) utilizando una ligasa;
(d) crear una biblioteca de moléculas de ácido nucleico amplificada por PCR de las moléculas de ácido nucleico que se obtienen en la etapa (c) utilizando un cebador capaz de hibridarse con el complemento de la molécula adaptadora 5 y un cebador capaz de hibridarse con la molécula adaptadora 3.
(e) secuenciar la biblioteca amplificada resultante de moléculas de ácido nucleico; y
(f) analizar los resultados de la secuenciación para determinar si la molécula de ácido nucleico está presente en la biblioteca de moléculas de ácido nucleico.
Preferentemente, el ácido nucleico diana se asocia con una enfermedad o estado de pre-enfermedad. Preferentemente, el ácido nucleico diana se asocia con un organismo en particular. Preferentemente, el ácido nucleico diana se asocia con un tipo de tejido particular. Preferentemente, el ácido nucleico diana se asocia con un estado de desarrollo particular.
En realizaciones preferidas de los aspectos descritos anteriormente, las moléculas de ácido nucleico son moléculas de... [Seguir leyendo]
Reivindicaciones:
1. Un método para reducirla tendencia de secuencia de una técnica de secuenciación que implica la unión con un adaptador, comprendiendo el método:
(a) proporcionar un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 3 bloqueados (moléculas adaptadoras 3) y un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 5 bloqueados (moléculas adaptadoras 5), donde las moléculas 3 y 5 comprenden uno o más nucleótidos degenerados;
(b) unir las moléculas adaptadoras 3 a los extremos 3 de las moléculas de ácido nucleico diana utilizando una
ligasa;
(c) unir las moléculas adaptadoras 5 a los extremos 5 de las moléculas de ácido nucleico diana de la etapa (b) utilizando una ligasa; y
(d) determinar la secuencia de las moléculas de ácido nucleico diana que se obtienen en la etapa (c) utilizando un cebador capaz de hibridarse con el complemento de la molécula adaptadora 5 y un cebador capaz de hibridarse con la molécula adaptadora 3.
2. Un método para detectar una molécula de ácido nucleico diana en un biblioteca de moléculas de ácidos nucleicos, comprendiendo el método:
(a) proporcionar un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 3 bloqueados (moléculas adaptadoras 3) y un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 5 bloqueados (moléculas adaptadoras 5), donde las moléculas 3 y 5 comprenden uno o más nucleótidos degenerados y se pueden unir a la molécula de ácido nucleico diana;
(b) unir la molécula adaptadora 3 a los extremos 3 de las moléculas de ácido nucleico de la biblioteca de moléculas de ácido nucleico utilizando una ligasa;
(c) unir la molécula adaptadora 5 a los extremos 5 de moléculas de ácido nucleico de la etapa (b) utilizando una ligasa;
(d) crear una biblioteca amplificada de moléculas de ácido nucleico por PCR de las moléculas de ácido nucleico que se obtienen en la etapa (c) utilizando un cebador capaz de hibridarse con el complemento de la molécula adaptadora 5 y un cebador capaz de hibridarse con la molécula adaptadora 3;
(e) secuenciar la biblioteca amplificada resultante de moléculas de ácido nucleico; y
(f) analizar los resultados de la secuenciación para determinar si la molécula de ácido nucleico diana está presente en la biblioteca de moléculas de ácido nucleico.
3. El método de acuerdo con la reivindicación 2, donde el ácido nucleico diana se asocia con una enfermedad o estado de pre-enfermedad.
4. El método de acuerdo con la reivindicación 2, donde el ácido nucleico diana se asocia con un organismo en particular.
5. El método de acuerdo con la reivindicación 2, donde el ácido nucleico diana se asocia con un tipo de tejido en particular.
6. El método de acuerdo con la reivindicación 2, donde el ácido nucleico diana se asocia con un estado de desarrollo en particular.
7. El método de acuerdo con una cualquiera de las reivindicaciones 1-6, donde las moléculas de ácido nucleico son moléculas de ARN.
8. El método de acuerdo con una cualquiera de las reivindicaciones 1-6, donde las moléculas de ácido nucleico son moléculas de ADN.
9. Un método para generar una biblioteca de ADNc a partir de una biblioteca de moléculas de ARN, comprendiendo el método:
(a) proporcionar un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 3 bloqueados (moléculas adaptadoras 3) y un grupo de oligonucleótidos de cadena sencilla de secuencia conocida con extremos 5 bloqueados (moléculas adaptadoras 5), donde las moléculas 3 y 5 comprenden uno o más nucleótidos degenerados;
(b) unir las moléculas adaptadoras 3 a los extremos 3 de las moléculas de ácido nucleico diana utilizando una ligasa;
(c) unir las moléculas adaptadoras 5 a los extremos 5 de las moléculas de ácido nucleico diana de la etapa (b) utilizando una ligasa;
(d) crear moléculas de estructura híbrida ARN/ADN a partir de moléculas de ARN que se obtienen en la etapa (c) utilizando una enzima transcriptasa inversa y un cebador capaz de hibridarse con la molécula adaptadora 3; y
(e) crear una biblioteca de ADNc por PCR de las moléculas de estructura híbrida ARN/ADN que se obtienen en la etapa (d) utilizando un cebador capaz de hibridarse con el complemento de la molécula adaptadora 5 y un cebador capaz de hibridarse con la molécula adaptadora 3.
5 10. Un grupo de ollgonucleótldos de cadena sencilla de secuencia conocida con extremos 3 bloqueados (moléculas
adaptadoras 3) y un grupo de ollgonucleótidos de cadena sencilla de secuencia conocida con extremos 5 bloqueados (moléculas adaptadoras 5), donde las moléculas adaptadoras 3 y 5 comprenden uno o más nucleótidos degenerados, para su uso en un método como se define en una cualquiera de las reivindicaciones 1-9.
Patentes similares o relacionadas:
Método y medios para purificar vectores retrovíricos, del 29 de Julio de 2020, de Autolus Limited: Una célula productora de retrovirus que expresa una proteína de marcaje en la superficie celular, de tal manera que los vectores retrovíricos producidos por la célula se […]
Biblioteca de péptidos y su uso, del 8 de Julio de 2020, de DAIICHI SANKYO COMPANY, LIMITED: Una biblioteca de péptidos que comprende una pluralidad de péptidos diferentes en la que los péptidos comprenden cada uno una secuencia de aminoácidos […]
Marcador de células endoteliales corneales, del 17 de Junio de 2020, de OSAKA UNIVERSITY: Método para producir una célula endotelial corneal, comprendiendo el método la etapa de clasificar, a partir de una población celular que comprende una célula […]
Vectores de AAV dirigidos a oligodendrocitos, del 10 de Junio de 2020, de THE UNIVERSITY OF NORTH CAROLINA AT CHAPEL HILL: Un ácido nucleico que codifica una cápside de AAV, comprendiendo el ácido nucleico una secuencia codificante de la cápside de AAV que es al menos el 96 % idéntica […]
Anticuerpo contra péptido codificado por exón-21 de periostina y composición farmacéutica para prevenir o tratar enfermedades asociadas a inflamación que contienen el mismo, del 6 de Mayo de 2020, de OSAKA UNIVERSITY: Anticuerpo que se une a uno o más péptidos seleccionados del grupo que consiste en un péptido codificado por el exón-21 de periostina que […]
Reconocimiento de unión a diana celular mediante un agente bioactivo usando transferencia de energía de resonancia de bioluminiscencia intracelular, del 6 de Mayo de 2020, de PROMEGA CORPORATION: Un sistema de ensayo que comprende: (a) una biblioteca de agentes bioactivos, cada uno de los cuales está fijado a un fluoróforo; (b) una diana celular fusionada a […]
Combinación de dos elementos genéticos para el control del desarrollo del tipo floral de una planta dicotiledónea, y utilización en procedimientos de detección y selección, del 1 de Abril de 2020, de Institut national de recherche pour l'agriculture, l'alimentation et l'environnement: Utilización de una combinación de dos elementos genéticos para el control del desarrollo del tipo floral de una planta dicotiledónea, comprendiendo dicha combinación, respectivamente: […]
Producción de proteínas en medios de cultivo celular libres de glutamina, del 25 de Marzo de 2020, de F. HOFFMANN-LA ROCHE AG: Un procedimiento para producir un polipéptido en una célula huésped de mamífero que expresa dicho polipéptido, que comprende cultivar la célula […]