Procedimiento para preparar derivados de quinazolina sustituida con aminocrotonilamino.
Procedimiento para preparar un compuesto de fórmula general
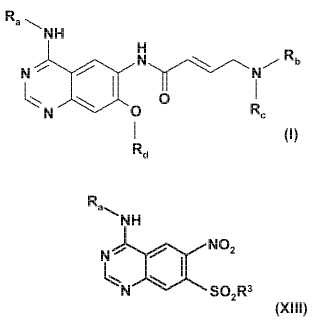
Ra indica un grupo bencilo,
1-feniletilo o 3-cloro-4-fluorofenilo,
Rb indica un grupo metilo, etilo, isopropilo, ciclopropilo, 2-metoxietilo, tetrahidrofuran-3-ilo, tetrahidrofuran-2-il-metilo,tetrahidrofuran-3-il-metilo, tetrahidropiran-4-ilo o tetrahidropiran-4-il-metilo,
Rc indica un grupo metilo, etilo o 2-metoxietilo o
Rb y Rc junto con el átomo de nitrógeno al que están unidos estos grupos, indican un grupo morfolino uhomomorfolino opcionalmente sustituido con uno o dos grupos alquilo C1-3, y
Rd indica un grupo ciclopropilmetilo, ciclobutilo, ciclopentilo, tetrahidrofuran-3-ilo, tetrahidrofuran-2-il-metilo,
tetrahidrofuran-3-il-metilo, tetrahidropiran-4-ilo o tetrahidropiran-4-il-metilo,
que comprende las etapas de procedimiento (realización A):
a) hacer reaccionar 7-cloro-6-nitro-3H-quinazolin-4-ona
con una amina primaria de fórmula Ra-NH2 (XV), en la que Ra indica un grupo bencilo, 1-feniletilo o 3-cloro-4-fluorofenilo, en presencia de POCI3,
b) convertir el compuesto resultante de fórmula general
en el derivado de sulfonilo de fórmula.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/EP2007/050752.
Solicitante: BOEHRINGER INGELHEIM INTERNATIONAL GMBH.
Nacionalidad solicitante: Alemania.
Dirección: BINGER STRASSE 173 55216 INGELHEIM AM RHEIN ALEMANIA.
Inventor/es: SCHROEDER, JUERGEN, DR., DZIEWAS,GEORG, FACHINGER,THOMAS, JAEGER,BURKHARD, REICHEL,CARSTEN, RENNER,SVENJA.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C07D293/04 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07D COMPUESTOS HETEROCICLICOS (Compuestos macromoleculares C08). › C07D 293/00 Compuestos heterocíclicos que contienen ciclos que tienen nitrógeno y selenio o nitrógeno y teluro, con o sin átomos de oxígeno o azufre, como heteroátomos del ciclo. › Ciclos de cinco miembros.
PDF original: ES-2397181_T3.pdf
Fragmento de la descripción:
Procedimiento para preparar derivados de quinazolina sustituida con aminocrotonilamino La invención se refiere a un procedimiento para preparar derivados de quinazolina sustituida con aminocrotonilamino de fórmula general (I)
en la que los grupos Ra, Rb, Rc y Rd tienen los significados dados en las reivindicaciones y en la memoria descriptiva.
Antecedentes de la invención Los derivados de quinazolina de fórmula general (I) se conocen de los documentos WO 02/50043 y WO 04/074263, que describen compuestos con propiedades farmacológicas valiosas, incluyendo en particular un efecto inhibidor de la transducción de señales mediada por tirosina quinasas y un efecto inhibidor de la transducción de señales mediada por el receptor del factor de crecimiento epidérmico (EGF-R) . Por lo tanto, los compuestos de este tipo son adecuados para el tratamiento de enfermedades, en particular para el tratamiento de enfermedades tumorales, enfermedades de los pulmones y las vías aéreas y enfermedades del tracto gastrointestinal y los conductos biliares y la vesícula biliar.
El documento WO 2002/50043 describe un método de producción en el que se preparan quinazolinas sustituidas con aminocrotonilamino (I) en una reacción en un solo recipiente, a partir del correspondiente componente de anilina (II) , ácido bromocrotónico, cloruro de oxalilo y una amina secundaria (véase el diagrama 1) .
Diagrama 1:
El procedimiento no es adecuado para el uso técnico a escala industrial, ya que los rendimientos obtenidos son como máximo de 50% y en general es necesaria una purificación laboriosa por cromatografía en columna. Además, el educto del ácido bromocrotónico no está disponible en el comercio en grandes cantidades y el correspondiente bromocrotonato de metilo solo está disponible con una pureza de aproximadamente 80%.
El documento WO 2005/037824 describe un procedimiento alternativo para preparar derivados de quinazolina sustituida con aminocrotonilamino de fórmula general (I) por reacción de Wittig-Horner-Emmons de quinazolinas sustituidas con dialquilfosfonoacetamido (III) con un 2-aminoacetaldehído (IV) (diagrama 2) , aunque en lugar del aldehído (IV) se puede usar el correspondiente hidrato o un acetal (p. ej., el acetal dietílico correspondiente a (IV) ) , a partir del cual se libera el aldehído (previamente o in situ) .
Diagrama 2:
Los eductos de fórmula III se pueden obtener como sigue según el documento WO 2005/037824: Diagrama 3:
Tanto en la técnica anterior descrita anteriormente como dentro del alcance de la invención descrita en lo sucesivo, Ra, Rb, Rc, Rd, R1 y R2 tienen los siguientes significados: Ra indica un grupo bencilo, 1-feniletilo o 3-cloro-4-fluorofenilo, Rb indica un grupo metilo, etilo, isopropilo, ciclopropilo, 2-metoxietilo, tetrahidrofuran-3-ilo, tetrahidrofuran-2-il-metilo, 10 tetrahidrofuran-3-il-metilo, tetrahidropiran-4-ilo o tetrahidropiran-4-il-metilo, Rc indica un grupo metilo, etilo o 2-metoxietilo o Rb y Rc junto con el átomo de nitrógeno al que están unidos estos grupos, indican un grupo morfolino u homomorfolino opcionalmente sustituido con uno o dos grupos alquilo C1-3, Rd indica un grupo ciclopropilmetilo, ciclobutilo, ciclopentilo, tetrahidrofuran-3-ilo, tetrahidrofuran-2-il-metilo, 15 tetrahidrofuran-3-il-metilo, tetrahidropiran-4-ilo o tetrahidropiran-4-il-metilo, y R1 y R2 cada uno independientemente del otro indican un grupo alquilo C1-4, por ejemplo cada uno indica un grupo etilo.
Por un grupo homomorfolino se entiende el siguiente homólogo de anillo más grande del grupo morfolino, en concreto el grupo de fórmula
.
Partiendo del ácido 4-cloro-antranílico (V) que se puede obtener en el comercio, la reacción con acetato de formamidina (etapa a) produce la quinazolina (VI) , que después se nitra usando ácido sulfúrico y ácido nítrico concentrado (etapa b) . El regioisómero (VII) deseado después se clora usando cloruro de tionilo en acetonitrilo (etapa c) y el producto de cloración (IX) se hace reaccionar in situ con la correspondiente amina Ra-NH2 (etapa d) . El compuesto de fórmula (X) así obtenido se hace reaccionar por sustitución nucleófila catalizada por base, con Rd-OH para formar el compuesto (XI) (etapa e) , que a su vez se convierte por hidrogenación en la correspondiente aminoquinazolina (XII) (etapa f) . La aminoquinazolina (XII) después por reacción con ácido di- (alquil C1-4) fosfonoacético, p. ej., con ácido dietilfosfonoacético, en disolventes adecuados tales como tetrahidrofurano (THF) , dimetilformamida (DMF) o acetato de etilo, después de la correspondiente activación, por ejemplo, con 1, 1carbonildiimidazol, 1, 1-carbonilditriazol o anhídrido propanofosfónico, se convierte en la quinazolina sustituida con dialquilfosfonoacetamido (III) necesaria para la reacción de Wittig-Horner-Emmons.
El procedimiento descrito en el documento WO 2005/037824 también tiene una serie de desventajas graves para el uso técnico. Por ejemplo, el uso de cloruro de tionilo en la etapa (c) es problemático por razones de seguridad. Los alcoholes cíclicos o heterocíclicos necesarios para introducir el grupo Rd en un exceso de aproximadamente 2 equivalentes (eq.) son materiales de partida que son difíciles de obtener o caros, y también es necesaria la catálisis de transferencia de fase, por ejemplo usando 18-corona-6, para hacerlos reaccionar según la etapa (e) en el
diagrama 3 a una escala industrial. El producto de reacción debe purificarse por recristalización para eliminar el catalizador de transferencia de fase. La hidrogenación de la etapa (f) se lleva a cabo con la adición de ácido acético, si el educto contiene un átomo de cloro, para así prevenir la formación de subproductos desclorados que son difíciles de separar. La adición de ácido acético hace que se disuelvan trazas de níquel necesario como catalizador, y este es arrastrado a la etapa final y da lugar a un problema de metal pesado que es inaceptable para uso farmacéutico. Además, la capacidad de las reacciones parciales individuales debe mejorar; por ejemplo, la capacidad en la etapa (e) según el diagrama 3 es solo de 1/60 (1 kg de material de partida requiere un volumen de reactor de 60 litros) .
A la luz de las desventajas del método de producción conocido descrito antes, el problema de la presente invención es proporcionar un método mejorado, adecuado para la síntesis a una escala industrial, que permita la preparación segura de derivados de quinazolina sustituida con aminocrotonilamino (I) usando materiales de partida que se puedan obtener fácilmente, de alta pureza y a un coste técnico menor.
Descripción detallada de la invención El problema expuesto antes se resuelve según la invención por el siguiente procedimiento para preparar un compuesto de fórmula general
en la que de Ra a Rd son como se han descrito en lo que antecede, que comprende las siguientes etapas (realización A) :
a) hacer reaccionar 7-cloro-6-nitro-3H-quinazolin-4-ona con una amina primaria de fórmula Ra-NH2 (XV) , en la que Ra indica un grupo bencilo, 1-feniletilo o 3-cloro-4fluorofenilo, en presencia de POCI3,
b) convertir el compuesto resultante de fórmula general
en el derivado de sulfonilo de fórmula en la que R3 indica un grupo alquilo C1-4 en el que los átomos de hidrógeno pueden estar total o parcialmente sustituidos por átomos de flúor, o
un grupo fenilo opcionalmente sustituido con uno a tres sustituyentes seleccionados de grupos alquilo C1-3, átomos de halógeno, en particular átomos de flúor, cloro o bromo, grupos ciano o nitro, donde los sustituyentes pueden ser iguales o diferentes, y donde Ra en las dos fórmulas (X) y (XIII) tiene el significado dado en a) ,
c) convertir el derivado de sulfonilo de fórmula (XIII) en un compuesto de fórmula
por reacción con un alcohol de fórmula Rd-OH (XVI) en presencia de una base,
en la que Ra tiene los significados dados en a) y Rd indica un grupo ciclopropilmetilo, ciclobutilo, ciclopentilo, tetrahidrofuran-3-ilo, tetrahidrofuran-2-il-metilo, tetrahidrofuran-3-il-metilo, tetrahidropiran-4-ilo o tetrahidropiran-4-ilmetilo,
d) reducir el compuesto de fórmula (XI) así obtenido al derivado amino de fórmula
en la que Ra tiene los significados dados en a) y Rd tiene los significados dados en c) , e) convertir los derivados amino de fórmula (XII) en el éster fosfónico de fórmula
en la que Ra tiene los significados dados en a) y Rd tiene los significados dados en c) , y R1 y R2 cada uno independientemente del otro indica un grupo alquilo C1-4, pero preferiblemente grupos etilo,
f) hacer reaccionar el éster fosfónico de fórmula (III) resultante con un... [Seguir leyendo]
Reivindicaciones:
1. Procedimiento para preparar un compuesto de fórmula general
Ra indica un grupo bencilo, 1-feniletilo o 3-cloro-4-fluorofenilo,
Rb indica un grupo metilo, etilo, isopropilo, ciclopropilo, 2-metoxietilo, tetrahidrofuran-3-ilo, tetrahidrofuran-2-il-metilo, tetrahidrofuran-3-il-metilo, tetrahidropiran-4-ilo o tetrahidropiran-4-il-metilo, Rc indica un grupo metilo, etilo o 2-metoxietilo o Rb y Rc junto con el átomo de nitrógeno al que están unidos estos grupos, indican un grupo morfolino u homomorfolino opcionalmente sustituido con uno o dos grupos alquilo C1-3, y
Rd indica un grupo ciclopropilmetilo, ciclobutilo, ciclopentilo, tetrahidrofuran-3-ilo, tetrahidrofuran-2-il-metilo, tetrahidrofuran-3-il-metilo, tetrahidropiran-4-ilo o tetrahidropiran-4-il-metilo, que comprende las etapas de procedimiento (realización A) : a) hacer reaccionar 7-cloro-6-nitro-3H-quinazolin-4-ona con una amina primaria de fórmula Ra-NH2 (XV) , en la que Ra indica un grupo bencilo, 1-feniletilo o 3-cloro-4fluorofenilo, en presencia de POCI3,
b) convertir el compuesto resultante de fórmula general
en el derivado de sulfonilo de fórmula en la que R3 indica un grupo alquilo C1-4 en el que los átomos de hidrógeno pueden estar total o parcialmente sustituidos por átomos de flúor, o
un grupo fenilo opcionalmente sustituido con uno a tres sustituyentes seleccionados de grupos alquilo C1-3, átomos de halógeno, en particular átomos de flúor, cloro o bromo, grupos ciano o nitro, donde los sustituyentes pueden ser iguales o diferentes, y donde Ra en las dos fórmulas (X) y (XIII) tiene los significados dados en a) ,
c) convertir el derivado de sulfonilo de fórmula (XIII) en un compuesto de fórmula por reacción con un alcohol de fórmula Rd-OH (XVI) en presencia de una base,
en la que Ra tiene los significados dados en a) y Rd indica un grupo ciclopropilmetilo, ciclobutilo, ciclopentilo, 10 tetrahidrofuran-3-ilo, tetrahidrofuran-2-il-metilo, tetrahidrofuran-3-il-metilo, tetrahidropiran-4-ilo o tetrahidropiran-4-ilmetilo,
d) reducir el compuesto de fórmula (XI) así obtenido al derivado amino de fórmula en la que Ra tiene los significados dados en a) y Rd tiene los significados dados en c) , 15 e) convertir los derivados amino de fórmula (XII) en el éster fosfónico de fórmula en la que Ra tiene los significados dados en a) y Rd tiene los significados dados en c) , R1 y R2 cada uno independientemente del otro indica un grupo alquilo C1-4,
f) hacer reaccionar el éster fosfónico de fórmula (III) resultante con un aducto de hidrogenosulfito de fórmula ,
en la que M+ indica un catión o un protón y
Rb indica un grupo metilo, etilo, isopropilo, ciclopropilo, 2-metoxietilo, tetrahidrofuran-3-ilo, tetrahidrofuran-2-il-metilo, tetrahidrofuran-3-il-metilo, tetrahidropiran-4-ilo o tetrahidropiran-4-il-metilo, Rc indica un grupo metilo, etilo o 2-metoxietilo o Rb y Rc junto con el átomo de nitrógeno al que están unidos estos grupos, indican un grupo morfolino u
homomorfolino opcionalmente sustituido con uno o dos grupos alquilo C1-3, en la forma de una reacción de Wittig-Horner-Emmons. 2. Uso del procedimiento según la reivindicación 1, para preparar el compuesto
4-