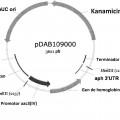
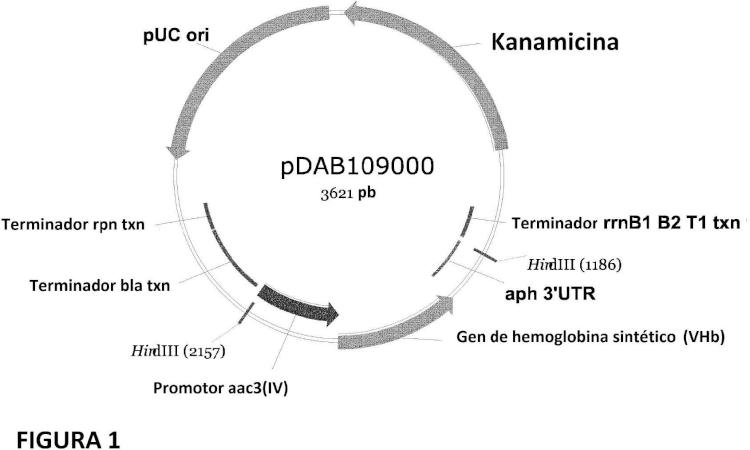
Mejora de la producción de espinosinas con proteínas que se unen al oxígeno.
Un método para aumentar los títulos de espinosinas de una célula hospedante de S.
spinosa, comprendiendo dicho método transformar la célula hospedante de S. spinosa con un gen heterólogo que codifica una proteína que se une al oxígeno.
Tipo: Patente Europea. Resumen de patente/invención. Número de Solicitud: E12166535.
Solicitante: DOW AGROSCIENCES LLC.
Nacionalidad solicitante: Estados Unidos de América.
Dirección: 9330 ZIONSVILLE ROAD INDIANAPOLIS, IN 46268 ESTADOS UNIDOS DE AMERICA.
Inventor/es: MOUNCEY,NIGEL, HAN,LEI.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C12N1/19 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12N MICROORGANISMOS O ENZIMAS; COMPOSICIONES QUE LOS CONTIENEN; PROPAGACION, CULTIVO O CONSERVACION DE MICROORGANISMOS; TECNICAS DE MUTACION O DE INGENIERIA GENETICA; MEDIOS DE CULTIVO (medios para ensayos microbiológicos C12Q 1/00). › C12N 1/00 Microorganismos, p.ej. protozoos; Composiciones que los contienen (preparaciones de uso médico que contienen material de protozoos, bacterias o virus A61K 35/66, de algas A61K 36/02, de hongos A61K 36/06; preparación de composiciones de uso médico que contienen antígenos o anticuerpos bacterianos, p. ej. vacunas bacterianas, A61K 39/00 ); Procesos de cultivo o conservación de microorganismos, o de composiciones que los contienen; Procesos de preparación o aislamiento de una composición que contiene un microorganismo; Sus medios de cultivo. › modificados por la introducción de material genético extraño.
- C12N15/52 C12N […] › C12N 15/00 Técnicas de mutación o de ingeniería genética; ADN o ARN relacionado con la ingeniería genética, vectores, p. ej. plásmidos, o su aislamiento, su preparación o su purificación; Utilización de huéspedes para ello (mutantes o microorganismos modificados por ingeniería genética C12N 1/00, C12N 5/00, C12N 7/00; nuevas plantas en sí A01H; reproducción de plantas por técnicas de cultivo de tejidos A01H 4/00; nuevas razas animales en sí A01K 67/00; utilización de preparaciones medicinales que contienen material genético que es introducido en células del cuerpo humano para tratar enfermedades genéticas, terapia génica A61K 48/00; péptidos en general C07K). › Genes que codifican enzimas o proenzimas.
- C12N15/90 C12N 15/00 […] › Introducción estable de ADN extraño en el cromosoma.
- C12P19/62 C12 […] › C12P PROCESOS DE FERMENTACION O PROCESOS QUE UTILIZAN ENZIMAS PARA LA SINTESIS DE UN COMPUESTO QUIMICO DADO O DE UNA COMPOSICION DADA, O PARA LA SEPARACION DE ISOMEROS OPTICOS A PARTIR DE UNA MEZCLA RACEMICA. › C12P 19/00 Preparación de compuestos que contienen radicales sacárido (ácido cetoaldónico C12P 7/58). › teniendo el heterociclo al menos ocho miembros y sólo oxígeno como heteroátomo del ciclo, p. ej. eritromicina, espiramicina, nistatina.
PDF original: ES-2553079_T3.pdf


Patentes similares o relacionadas:
 Animales no humanos que tienen un locus de cadena ligera lambda de inmunoglobulina modificado por ingeniería, del 29 de Julio de 2020, de REGENERON PHARMACEUTICALS, INC.: Un roedor cuyo genoma de la línea germinal comprende un locus de cadena ligera λ de inmunoglobulina endógeno que comprende:
(a) uno o más segmentos […]
Animales no humanos que tienen un locus de cadena ligera lambda de inmunoglobulina modificado por ingeniería, del 29 de Julio de 2020, de REGENERON PHARMACEUTICALS, INC.: Un roedor cuyo genoma de la línea germinal comprende un locus de cadena ligera λ de inmunoglobulina endógeno que comprende:
(a) uno o más segmentos […]
Métodos y composiciones para el tratamiento de enfermedades por almacenamiento lisosomal, del 27 de Mayo de 2020, de Sangamo Therapeutics, Inc: Uno o más transgenes y una o más nucleasas con dedos de zinc (ZFN) para su uso en un método de tratamiento de una enfermedad por almacenamiento lisosomal, el método […]
Terapias de aumento génico de la degeneración retiniana causada por mutaciones en el gen PRPF31, del 6 de Mayo de 2020, de MASSACHUSETTS EYE & EAR INFIRMARY: Un vector de virus adeno-asociado de tipo 2 (AAV2) que comprende una secuencia que codifica PRPF31 humano, operativamente enlazado a un promotor […]
Proteínas que tienen actividad nucleasa, proteínas de fusión y usos de estas, del 18 de Marzo de 2020, de HELMHOLTZ ZENTRUM MUNCHEN DEUTSCHES FORSCHUNGSZENTRUM FUR GESUNDHEIT UND UMWELT (GMBH): Una molécula de ácido nucleico que codifica (I) un polipéptido que tiene la actividad de una endonucleasa, que es (a) una molécula de ácido nucleico que […]
Animales no humanos que tienen una interrupción en un locus C9ORF72, del 19 de Febrero de 2020, de REGENERON PHARMACEUTICALS, INC.: Un roedor que comprende en su genoma una deleción de la porción codificante del exón 2 hasta la porción codificante del exón 11 de un locus C9orf72 endógeno, […]
Métodos y composiciones para modificar un locus objetivo, del 12 de Febrero de 2020, de REGENERON PHARMACEUTICALS, INC.: Un método para la modificación en serie de un locus objetivo en una célula, que comprende: (a) proporcionar la célula que comprende el locus objetivo, en donde el locus objetivo […]
Modelo de cerdo para la diabetes, del 29 de Enero de 2020, de AARHUS UNIVERSITET: Un cerdo transgénico que comprende un gen de polipéptido amiloide de los islotes (IAPP) mutado humano o parte del mismo, y que muestra al menos […]
Procedimientos de modificación de células hospedadoras, del 22 de Enero de 2020, de GLAXOSMITHKLINE BIOLOGICALS S.A.: Una célula hospedadora procariota aislada, en la que i) un gen heterólogo que codifica una oligosacaril transferasa y ii) ADN de al menos 8 kb que codifica […]