36 patentes, modelos y diseños de QUIMICA SINTETICA, S.A.
Procedimiento para la preparación de (1S,2R)-milnaciprán.
Sección de la CIP Química y metalurgia
(26/06/2019). Inventor/es: BARRECA, GIUSEPPE, ROMANO',BRUNO GAETANO. Clasificación: C07C231/00, C07C233/00.
Un procedimiento para la preparación de levomilnaciprán ((1S,2R)-2-(aminometil)-N,N-dietil-1- fenilciclopropanocarboxamida) que comprende las siguientes etapas:
a) convertir directamente la forma enantioméricamente enriquecida del alcohol (D) en la forma enantioméricamente enriquecida del derivado ftalimido (C) mediante tratamiento con ftalimida en presencia de una trialquil- o triarilfosfina y de un azodicarboxilato de dialquilo: **Fórmula**
en el que la cantidad de ftalimida está comprendida entre 1 y 1,3 equivalentes con respecto a la cantidad molar de alcohol (D) usada, y las cantidades de fosfina y de azodicarboxilato están comprendidas, independientemente entre sí, entre 1 y 1,5 equivalentes con respecto a la cantidad molar de alcohol (D) usada;
b) desbloquear la forma enantioméricamente enriquecida del derivado ftalimido (C) para obtener levomilnaciprán:**Fórmula**.
PDF original: ES-2746081_T3.pdf
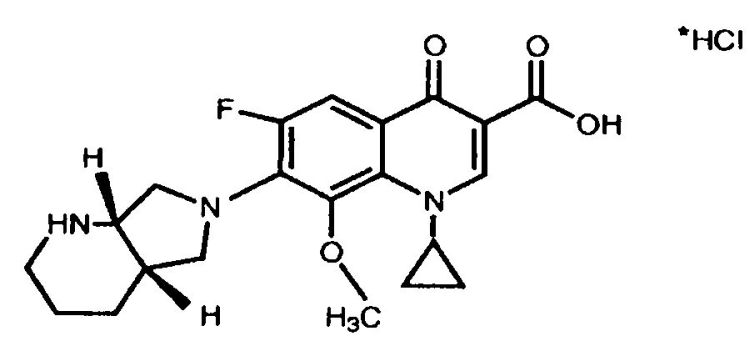
NUEVA FORMA CRISTALINA DE MOXIFLOXACINO CLORHIDRATO ANHIDRO FORMA IV.
(09/02/2010) Nueva forma cristalina de moxifloxacino clorhidrato anhidro forma IV.
La presente invención se refiere a una nueva forma polimórfica estable de moxifloxacino clorhidrato anhidro, a un procedimiento para la preparación de dicha forma polimórfica, y a composiciones farmacéuticas que comprenden dicha forma polimórfica
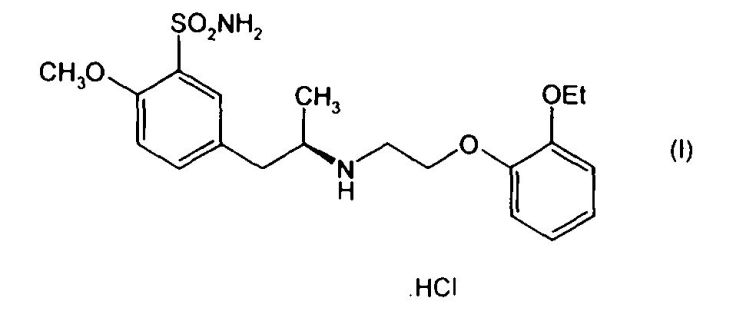
PROCEDIMIENTO PARA LA PREPARACION DE VALSARTAN.
(09/02/2010) La presente invención se refiere a un nuevo procedimiento para la preparación de valsartán a partir de un compuesto de fórmula general (I) en donde R es el grupo cumilo, tritilo o t-butilo.
El procedimiento consta de una primera etapa en la que se procede a la eliminación del grupo protector y a continuación se procede a la apertura del anillo de oxazolidinona por hidrogenación catalítica en presencia de una base orgánica.
Finalmente se aísla valsartán o una de sus sales farmacéuticamente aceptables
PROCEDIMIENTO PARA LA PREPARACION DE CARDESARTAN CILEXETILO EN FORMA CRISTALINA.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(22/12/2009). Inventor/es: COSME GOMEZ,ANTONIO, PALOMO NICOLAU,FRANCISCO, GONZALEZ HERNANDEZ,PEDRO, VICIOSO SANCHEZ, MERCEDES, RODRIGUEZ SALGADO,NATIVIDAD. Clasificación: A61K31/4184, C07D403/10, A61P9/12.
Procedimiento para la preparación de candesartán cilexetilo en forma cristalina.
Se describe un procedimiento para la preparación de una forma cristalina de candesartán cilexetilo denominada forma I que tiene un contenido de la denominada forma II inferior al 1% en peso. Dicho procedimiento comprende una etapa de siembra con un producto cristalino que comprende una proporción mayoritaria de la forma I, y una etapa de digestión. La invención también se refiere a dicha forma cristalina, a las composiciones farmacéuticas que la contienen, y a su uso para la preparación de un medicamento para el tratamiento de la hipertensión. Se ha observado que el candesartán cilexetilo forma I presenta una buena estabilidad cuando se mantiene en condiciones de estabilidad acelerada si presenta un contenido de la forma II inferior al 1 % en peso.
FORMA CRISTALINA DE MOXIFLOXACINO BASE.
Secciones de la CIP Química y metalurgia Necesidades corrientes de la vida
(22/12/2009). Inventor/es: VILLASANTE PRIETO,JAVIER, PALOMO NICOLAU,FRANCISCO. Clasificación: C07D471/04, A61K31/4709, A61P31/04.
Forma cristalina de moxifloxacino base.
La presente invención se refiere a una forma cristalina de moxifloxacino base, un procedimiento para la preparación de dicha forma cristalina, composiciones farmacéuticas que comprenden dicha forma cristalina, así como al uso de dicha forma cristalina para la preparación de un medicamento para tratamientos antibacterianos.
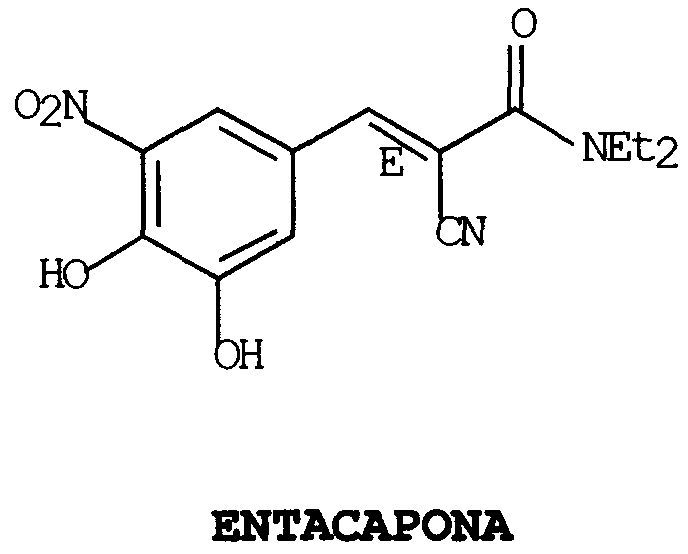
PROCEDIMIENTO PARA LA OBTENCION DE ENTACAPONA SUSTANCIALMENTE LIBRE DE SISOMERO, Z, SUS INTERMEDIOS DE SINTESIS Y NUEVA FORMA CRISTALINA.
Secciones de la CIP Química y metalurgia Necesidades corrientes de la vida
(11/12/2009). Inventor/es: SOLA CARANDELL,LLUIS, BENET-BUCHHOLZ, JORDI, DR., MOLINA PONCE,ANDRES, PALOMO NICOLAU,FCO. EUGENIO. Clasificación: C07C255/41, A61K31/277, C07C253/30, C07C235/34, A61P25/16.
Procedimiento para la obtención de entacapona sustancialmente libre de isómero Z, sus intermedios de síntesis y una nueva forma cristalina.
La presente invención se refiere a un nuevo procedimiento para la obtención de Entacapona sustancialmente libre de isómero Z a partir de 3,4-dihidroxi-5-nitrobenzaldehido y N,N-dimetil cianoacetamida, o bien, directamente a partir de una mezcla de isómeros E/Z de Entacapona, mediante la formación de sales orgánicas o inorgánicas, especialmente de piperidina y sodio. A partir de este procedimiento puede obtenerse una nueva forma cristalina de Entacapona G de manera rápida, eficaz, sencilla y sustancialmente libre de isómero Z. También es objeto de la invención una composición farmacéutica que la contenga.
PROCEDIMIENTO PARA LA PREPARACION DE MOXIFLOXACINO Y MOXIFLOXACINO CLORHIDRATO.
(19/08/2009) Procedimiento para la preparación de moxifloxacino, caracterizado por el hecho de que se lleva a cabo, en una reacción "one pot", la reacción del compuesto de fórmula (II)** ver fórmula** con un agente agente sililante de fórmula ZSiR3, en el que Z es Me3SiNH- o Cl-, y R es indistintamente una cadena alquílica C 1-4 o fenilo, en el seno de un disolvente polar aprótico, para obtener el compuesto intermedio de fórmula (VI), ** ver fórmula** que sin ser aislado se hace reaccionar a continuación con un compuesto trifluoruro de boro para obtener el compuesto intermedio de fórmula (VII) ** ver fórmula**, que no se aísla y se hace reaccionar posteriormente con (S,S)-2,8-diaza-biciclo[4.3.0]nonano(III), después de ajustar el pH a un valor comprendido entre 8 y 9, obteniéndose el compuesto intermedio (VIII)**…
NUEVA FORMA CRISTALINA DE ENTACAPONA Y PROCEDIMIENTO PARA SU OBTENCION.
Secciones de la CIP Química y metalurgia Necesidades corrientes de la vida
(07/08/2009). Ver ilustración. Inventor/es: SOLA CARANDELL,LLUIS, BENET-BUCHHOLZ, JORDI, DR., MOLINA PONCE,ANDRES, GARCIA RUIZ,GERMAN, PALOMO NICOLAU,FCO. EUGENIO. Clasificación: C07C255/41, A61P25/16, A61K31/277.
Nueva forma cristalina de entacapona y procedimiento para su obtención.#La invención se refiere a una nueva forma cristalina estable de entacapona, denominada Forma F. También se refiere al procedimiento para su obtención y a la composición farmacéuticamente aceptable que la comprende.#El procedimiento comprende disolver en caliente el producto entacapona en el seno de un disolvente orgánico; añadir la disolución obtenida en la etapa anterior sobre un anti-disolvente calentado previamente; enfriar la suspensión obtenida; y aislar el precipitado obtenido por filtración y, a continuación, secar el producto obtenido hasta peso constante.
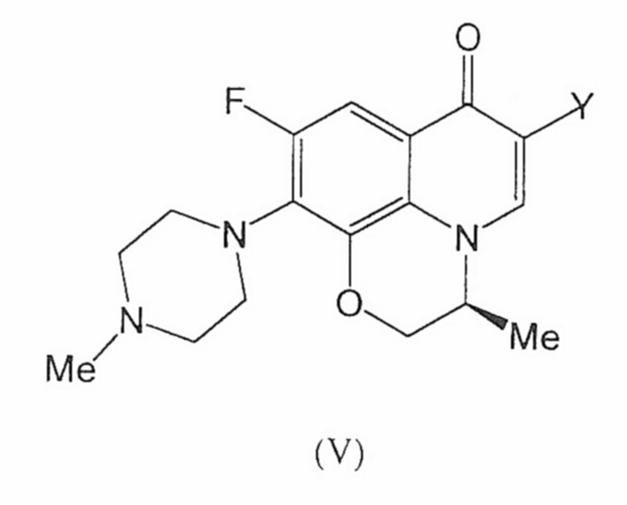
PROCEDIMIENTO PARA LA OBTENCION DE LEVOFLOXACINO EXENTO DE SALES.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(16/04/2009). Ver ilustración. Inventor/es: COSME GOMEZ,ANTONIO, PALOMO NICOLAU,FRANCISCO, MARTIN SANCHEZ,RAFAEL. Clasificación: A61P31/04, C07D498/06, A61K31/5383.
Procedimiento para obtener levofloxacino conteniendo no más del 0,1% en sales, que comprende la hidrólisis del compuesto (V), en presencia de una base, en el que Y es indistintamente un grupo COOR1, en el que R1 es un C1-C6 alquilo, o es un grupo BZ2 en el que Z representa un átomo de halógeno, un grupo C1-C6 alcoxi o un grupo C2-C7 alquilcarboniloxi, o bien Y es un grupo nitrilo, en el seno de una mezcla agua-alcohol C1-C4, posterior neutralización, separación de las sales, seguido de aislamiento de levofloxacino, sin la necesidad de llevar a cabo una etapa final de extracción y sin la utilización de disolventes halogenados.
PROCEDIMIENTO PARA LA PREPARACION DE VALACICLOVIR.
Sección de la CIP Química y metalurgia
(16/02/2009). Ver ilustración. Inventor/es: COSME GOMEZ,ANTONIO, VILLASANTE PRIETO,JAVIER, PALOMO NICOLAU,FRANCISCO, CEBALLOS MARRUECOS,EVA MARIA. Clasificación: C07C229/10, C07D473/18.
Procedimiento para la preparación de valaciclovir.#La presente invención se refiere a un procedimiento para la preparación de valaciclovir y sus sales farmacéuticas en el que se hace reaccionar un derivado activado del aciclovir con una enamina del aminoácido L-valina de fórmula general (II) que tiene el grupo carboxilo del aminoácido salificado con una base seleccionada entre el grupo formado por compuestos de amonio cuaternario, amidinas bicíclicas, amidinas lineales y fosfacenos. También se describen dichas enaminas del aminoácido L-valina, que son compuestos nuevos, y un procedimiento para obtenerlas.
SAL DE L-ARGININA DE ESOMEPRAZOL, PROCEDIMIENTO DE PREPARACION Y COMPOSICIONES FARMACEUTICAS QUE LA COMPRENDEN.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(16/12/2008). Ver ilustración. Inventor/es: PALOMO NICOLAU,FRANCISCO, MOLINA PONCE,ANDRES, PASTOR DEL CASTILLO,ALFREDO. Clasificación: A61K31/4439, C07D401/12, A61P1/04.
Sal de L-arginina de esomeprazol, procedimiento de preparación y composiciones farmacéuticas que la comprenden.#La presente invención se refiere a una nueva sal de esomeprazol, más específicamente a la sal de L-arginina de esomeprazol en forma física y químicamente estable, al procedimiento de su preparación y a las composiciones farmacéuticas que la comprenden.#El procedimiento comprende a) tratar esomeprazol con L-Arginina en el seno de un disolvente orgánico adecuado, b) concentrar y/o precipitar la sal deseada con un disolvente orgánico y c) aislar y secar la sal L-arginina de esomeprazol obtenida.
NUEVO PROCEDIMIENTO DE SITENSIS DE (6-(4-FENILBUTOXI)HEXIL)AMINAS N-S USTITUIDAS Y SU USO EN LA SINTESIS DE SALMETEROL XINAFOATO.
Sección de la CIP Química y metalurgia
(01/12/2008). Ver ilustración. Inventor/es: PALOMO NICOLAU,FRANCISCO, MOLINA PONCE,ANDRES, GARCIA RUIZ,GERMAN. Clasificación: C07C217/44.
Nuevo procedimiento de síntesis de [6-(4-fenilbutoxi)hexil]aminas N-sustituidas y su uso en la síntesis de salmeterol xinafoato.#La presente invención se refiere a un nuevo procedimiento para la preparación de los compuestos de fórmula general II y/o sus sales farmacéuticamente aceptables, donde R1 es un grupo susceptible de ser eliminado mediante hidrogenación.#Estos compuestos de fórmula II son útiles como intermedios en la síntesis de salmeterol y salmeterol xinafoato.
PROCEDIMIENTO PARA LA PREPARACION DE MOXIFLOXACINO Y MOXIFLOXACINO CLORHIDRATO.
Sección de la CIP Química y metalurgia
(01/11/2008). Ver ilustración. Inventor/es: COSME GOMEZ,ANTONIO, VILLASANTE PRIETO,JAVIER, PALOMO NICOLAU,FRANCISCO, MOLINA PONCE,ANDRES, FERNANDEZ LLORCA,SARA PENELOPE. Clasificación: C07D401/04, C07D215/56.
Procedimiento para la preparación de Moxifloxacino y Moxifloxacino Clorhidrato.#La presente invención se refiere a un procedimiento "one pot" para la preparación de moxifloxacino y su sal clorhidrato, sin la necesidad de aislar los compuestos intermedios.#El nuevo procedimiento permite la obtención del producto final moxifloxacino a gran escala con un buen rendimiento y en forma pura utilizando un procedimiento "one pot" en el que se utiliza menor cantidad de amina bicíclica, reactivo de alto coste. Además, con el presente procedimiento se evita y disminuye los tiempos y costes derivados del aislamiento de los intermedios y salva sustancialmente la obtención de la impureza que proviene de la desmetilación en la posición 8 del anillo.
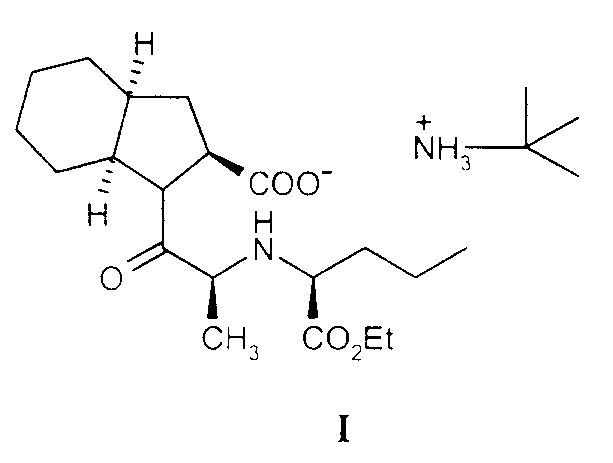
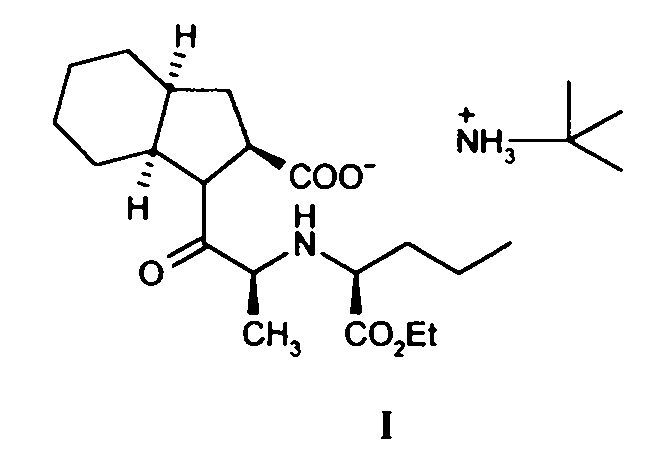
PROCEDIMIENTO PARA LA PREPARACION DE PERINDOPRIL ERBUMINA.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(01/11/2008). Ver ilustración. Inventor/es: PALOMO NICOLAU,FRANCISCO, DE LEON,DORCAS. Clasificación: A61K31/404, A61P9/12, C07D209/42, C07K5/02.
Procedimiento de obtención del ácido 1-[(2S)-2-[[(1S)-1(etoxicarbonil)butil]amino]-1-propionil] (2S,3aS,7aS)octahidro-1H-indol-2-carboxílico, sal de tert-butilamina que comprende hacer reaccionar un éster activo de N-[2(S)- (etoxicarbonil)butil]-L-alanina con una sal orgánica del ácido perhidroindol-2-carboxílico, seguido de adición de t-butilamina y de un ácido al medio de reacción, en cualquier orden, o bien de una sal formada entre un ácido y la terbutilamina y separar el producto por métodos convencionales.
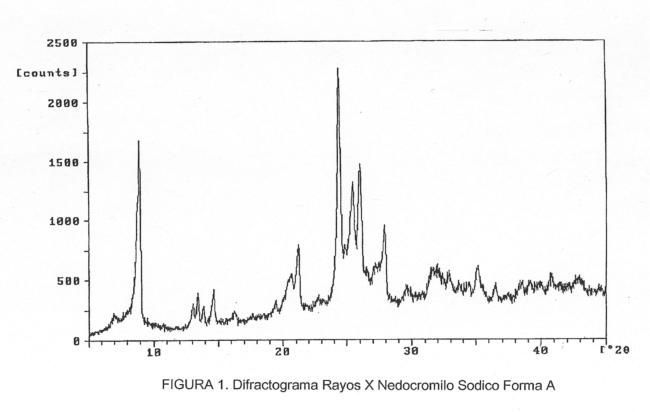
PROCEDIMIENTO PARA LA PREPARACION DE LAS FORMAS A Y C DE NEDROCROMILO SODICO.
Secciones de la CIP Química y metalurgia Necesidades corrientes de la vida
(16/06/2008). Ver ilustración. Inventor/es: PALOMO NICOLAU,FRANCISCO, DE LEON DE LEON,DORCAS. Clasificación: C07D491/04, A61K31/4741, C07D491/052.
Procedimiento para la preparación de las formas A y C de nedocromilo sódico.#La presente invención se refiere a un procedimiento para la preparación selectiva del trihidrato (Forma A) y del heptahemihidrato (Forma C) del nedocromilo sódico.
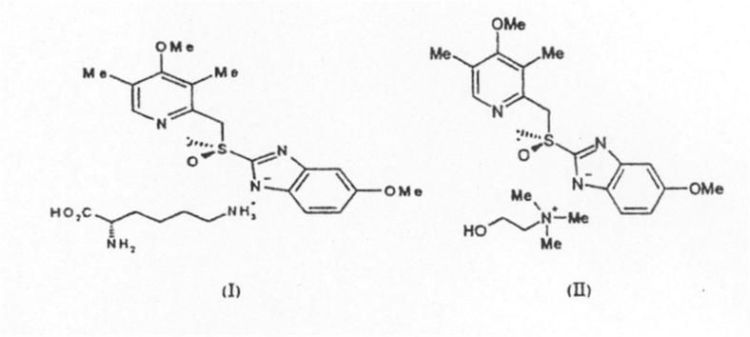
NUEVAS SALES DE ESOMEPRAZOL. PROCEDIMIENTO DE PREPARACION Y COMPOSICIONES FARMACEUTICAS QUE LAS COMPRENDEN.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(16/06/2008). Ver ilustración. Inventor/es: PALOMO NICOLAU,FRANCISCO, MOLINA PONCE,ANDRES, PASTOR DEL CASTILLO,ALFREDO. Clasificación: A61K31/4439, C07D401/12, C07D235/28, C07D213/68.
Nuevas sales de esomeprazol, procedimiento de preparación y composiciones farmacéuticas que las comprenden.#La presente invención se refiere a nuevas sales de esomeprazol, en particular se refiere a la sal de L-lisina de esomeprazol y la sal de colinade esomeprazol, de fórmulas (I) y (II) respectivamente. También es objeto de la invención proporcionar un procedimiento de preparación de dichas sales en forma estable y las composiciones farmacéuticas que las comprendan.
PROCEDIMIENTO PARA LA PREPARACION DE PANTOPRAZOL.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(01/04/2008). Ver ilustración. Inventor/es: PALOMO NICOLAU,FRANCISCO, MOLINA PONCE,ANDRES. Clasificación: A61K31/4439, C07D401/12, C07D235/28, C07D213/68.
Procedimiento para la preparación de pantoprazol.#La presente invención se refiere a un nuevo procedimiento para la obtención de pantoprazol (I) y sus sales farmacéuticamente aceptables#**FIGURA 01**#con un buen rendimiento y una elevada pureza. En dicho procedimiento se efectúa la introducción del grupo metoxi en la posición 4 del anillo de piridina del compuesto (IV)#**FIGURA 02**#por sustitución del átomo de cloro, mediante la reacción con un metóxido alcalino en el seno de una mezcla de metanol y un disolvente polar aprótico. También se refiere a un procedimiento de aislamiento que incluye un método para la purificación de pantoprazol, y a un procedimiento para la preparación del compuesto (IV), que se emplea como producto de partida en la preparación de pantoprazol.
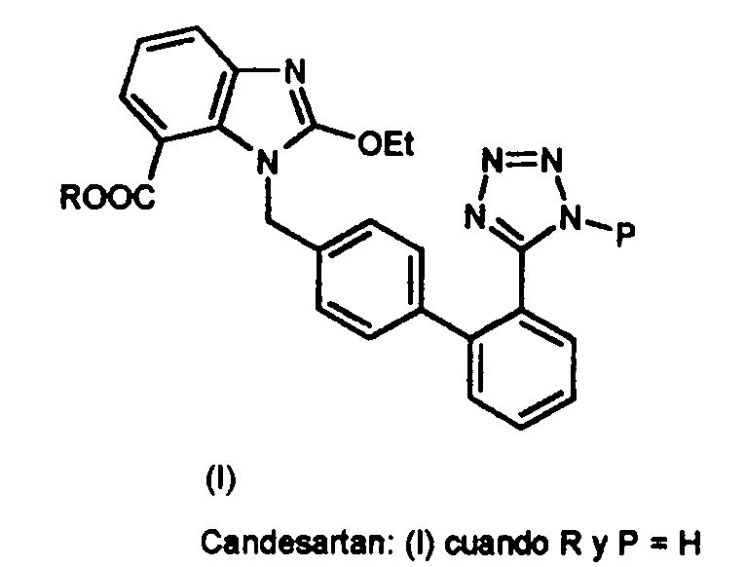
PROCEDIMIENTO PARA LA OBTENCION DE DERIVADOS DE BENCIMIDAZOL Y SUS INTERMEDIOS.
Sección de la CIP Química y metalurgia
(01/03/2008). Ver ilustración. Inventor/es: COSME GOMEZ,ANTONIO, PALOMO NICOLAU,FRANCISCO, VICIOSO SANCHEZ, MERCEDES. Clasificación: C07D235/26, C07D403/10.
Procedimiento para la obtención de derivados de bencimidazol y sus intermedios.#La presente invención se refiere a un procedimiento para la obtención de derivados de bencimidazol y sus intermedios, preferentemente, para la obtención de Candesartan y Candesartan cilexetilo.#Dicho procedimiento permite obtener los derivados de bencimidazol con un mejor rendimiento.
PROCEDIMIENTO ENZIMATICO PARA LA PREPARACION DE UN COMPUESTO INTERMEDIO Y SU USO EN LA SINTESIS DE TAMSULOSINA CLORHIDRATO.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(01/12/2007). Ver ilustración. Inventor/es: VILLASANTE PRIETO,JAVIER, PALOMO NICOLAU,FRANCISCO. Clasificación: A61K31/18, C07C231/02, C07C231/18, C12P13/02, C07C311/37, C07C233/22, C07C303/36, C12P41/00D2, C12P41/00.
Procedimiento enzimático para la preparación de un compuesto intermedio y su uso en la síntesis de tamsulosina clorhidrato.#La presente invención se refiere a un procedimiento de acilación enzimático enantioselectivo, para la preparación de compuestos (R)-2-halo-N-[2-(4-metoxi fenil)-1-metil-etil]acetamida, de fórmula III, en donde X= Cl, Br, I.#Estos compuestos III son útiles como intermedios en la síntesis de tamsulosina clorhidrato.
PROCEDIMIENTO PARA LA OBTENCION DE LEVOFLOXACINO EXENTO DE SALES.
Sección de la CIP Química y metalurgia
(16/08/2007). Ver ilustración. Inventor/es: COSME GOMEZ,ANTONIO, PALOMO NICOLAU,FRANCISCO, MARTIN SANCHEZ,RAFAEL. Clasificación: C07D498/06.
Procedimiento para la obtención de levofloxacino exento de sales.#La presente invención se refiere a un procedimiento para la obtención de levofloxacino exento de sales. En dicho procedimiento se emplea como producto de partida el compuesto (V) cuya hidrólisis alcalina en el seno de una mezcla agua-alcohol C1-C4 posterior neutralización y separación de las sales, conduce a levofloxacino exento de sales.#Una característica del procedimiento descrito es que no son necesarias extracciones en la etapa final del proceso.
PROCEDIMIENTO PARA LA PREPARACION DE PERINDOPRIL ERBUMINA.
Sección de la CIP Química y metalurgia
(16/08/2007). Ver ilustración. Inventor/es: PALOMO NICOLAU,FRANCISCO, DE LEON,DORCAS. Clasificación: C07D209/42.
Procedimiento para la preparación de perindopril erbumina.#La presente invención se refiere a un nuevo procedimiento para la preparación de perindopril erbumina (I), útil para el tratamiento de la hipertensión. La invención se refiere a la obtención del ácido 1-[(2S)-2-[[(1S)-1(etoxicarbonil)butil]amino]-1-propionil](2S,3aS,7aS)-octahidro-1H-indol-2-carboxílico, sal de tert-butilamina que comprende hacer reaccionar un éster activo de N-[2(S)-(etoxicarbonil)butil]-L-alanina con una sal orgánica del ácido perhidroindol-2-carboxílico, seguido de adición de t-butilamina y de un ácido al medio de reacción, en cualquier orden, o bien de una sal formada entre un ácido y la terbutilamina y separar el producto por métodos convencionales.
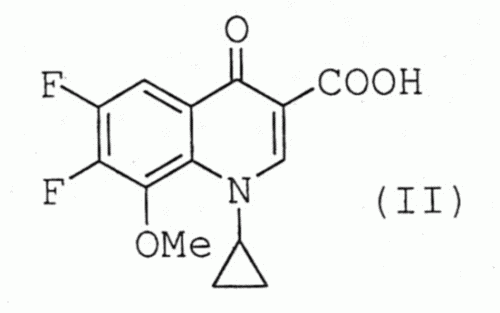
PROCEDIMIENTO SIMPLIFICADO PARA LA OBTENCION DE GATIFLOXACINO.
Secciones de la CIP Química y metalurgia Necesidades corrientes de la vida
(01/08/2006). Ver ilustración. Inventor/es: COSME GOMEZ,ANTONIO, VILLASANTE PRIETO,JAVIER, PALOMO NICOLAU,FRANCISCO. Clasificación: C07D401/04, A61P31/00, A61K31/496.
Procedimiento simplificado para la obtención de gatifloxacino. La presente invención se refiere a un procedimiento simplificado para la obtención de gatifloxacino. En dicho procedimiento se emplea como producto de partida el compuesto (II) que se hace reaccionar con 2-metilpiperazina después de ser sililado y activado en forma de quelato de boro. Finalmente, se elimina el quelato de boro por tratamiento con un alcohol de cadena alquílica C1-C4. Una característica del procedimiento descrito es que todas las reacciones se efectúan sin aislar los compuestos intermedios formados (procedimiento "one pot").
FORMULA CRISTALINA NO HIGROSCOPICA DE GATIFLOXACINO.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(01/08/2006). Inventor/es: COSME GOMEZ,ANTONIO, VILLASANTE PRIETO,JAVIER, PALOMO NICOLAU,FRANCISCO. Clasificación: A61P31/04, C07D401/04, A61K31/496.
Forma cristalina no higroscópica de gatifloxacino. La presente invención se refiere a una forma cristalina no higroscópica de gatifloxacino que contiene entre el 8 y el 9 % en peso de agua, a un procedimiento para obtenerla y a su uso como principio activo en la preparación de formulaciones farmacéuticas.
FORMA CRISTALINA DE GATIFLOXACINO.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(01/08/2006). Ver ilustración. Inventor/es: COSME GOMEZ,ANTONIO, VILLASANTE PRIETO,JAVIER, PALOMO NICOLAU,FRANCISCO. Clasificación: A61P31/04, C07D401/04, A61K31/496.
Forma cristalina de gatifloxacino. La presente invención se refiere a una forma cristalina de gatifloxacino obtenible por un procedimiento que comprende la recristalización del gatifloxacino crudo en metanol y que se estabiliza con un contenido en agua comprendido entre el 2,5 y el 4,5 % en peso, a un procedimiento para obtenerla y a su uso como principio activo en la preparación de formulaciones farmacéuticas.
PREPARACION DE POLIMORFOS CRISTALINOS DE FOSINOPRIL SODICO.
(01/12/2005) Un procedimiento para la preparación selectiva de los polimorfos cristalinos de la sal sódica del fosinopril que se caracteriza por formar una disolución total o parcial de fosinopril sódico en un disolvente o mezcla de disolventes que contiene menos de un 0, 2% (v/v) de agua respecto del total de agua más disolvente, de manera que: (a) el disolvente se selecciona de entre los que contienen oxígeno en su molécula y los nitrilos o mezcla de los mismos, con la condición de que el disolvente o mezcla de disolventes contenga más de un 0, 4% (v/v) de un alcohol alifático C1-C4, lineal o ramificado, en cuyo caso el polimorfo A se cristaliza a una temperatura comprendida entre 0ºC y 50ºC y se separa de la mezcla,…
SALES DE ADICION DE AZITROMICINA Y ACIDO CITRICO Y PROCEDIMIENTO PARA SU OBTENCION.
Sección de la CIP Química y metalurgia
(16/10/2005). Ver ilustración. Inventor/es: COSME GOMEZ,ANTONIO, PALOMO NICOLAU,FRANCISCO EUGE. Clasificación: C07H17/08.
Sales de adición de azitromicina y ácido cítrico y procedimiento para su obtención. Dichas sales de adición tienen una relación molar entre azitromicina y ácido cítrico tal que proporciona un pH, en una disolución acuosa al 10%, comprendido entre 4,0 y 8,0. El procedimiento para preparar dichas sales comprende: a) disolver azitromicina en un disolvente o mezcla de disolventes, b) añadir ácido cítrico, y c) aislar el producto obtenido la sal por cristalización. Las sales de adición de azitromicina y ácido cítrico son estables y solubles en medio acuoso, siendo útiles agentes antibacterianos y antiprotozoarios.
PROCEDIMIENTO PARA LA PREPARACION DE POLIMORFOS CRISTALINOS DE LA SAL SODICA DEL FOSINOPRIL.
Sección de la CIP Química y metalurgia
(16/10/2004). Inventor/es: PALOMO NICOLAU,FRANCISCO, GALLEGO PATO, SANDRA, PALOMO COLL, ANTONIO,LUIS. Clasificación: C07F9/572.
Procedimiento para la preparación de polimorfos cristalinos de la sal sódica del fosinopril. Se describe un nuevo procedimiento para la preparación selectiva de los polimorfos cristalinos A y B del fosinopril, especialmente del polimorfo A. El procedimiento descrito permite evitar el empleo de cantidades significativas de agua, lo que redunda en un riesgo menor de degradaciones hidrolíticas del principio activo.
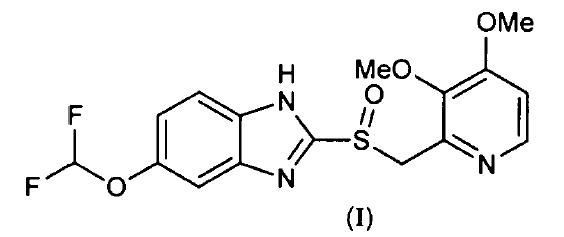
NUEVO PROCEDIMIENTO DE OBTENCION DE DERIVADOS DE 2-(2-PIRIDINILMETILSULFINIL)-1H-BENZIMIDAZOL.
Sección de la CIP Química y metalurgia
(16/01/2004). Ver ilustración. Inventor/es: COSME,ANTONIO, FAU DE CASA-JUANA,MIGUEL, GELPI,JOSE MARIA, MARTIN,RAFAEL, MOLINA,ANDRES. Clasificación: C07D401/12.
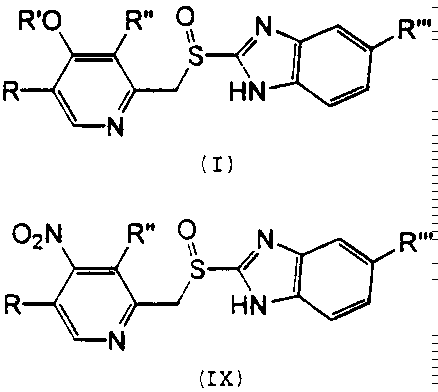
Nuevo procedimiento de obtención de derivados de 2-(2- piridinilmetilsulfinil)-1h-benzimidazol. Procedimiento de obtención de derivados de 2-(2-piridinilmetilsulfinil)-1H- benzimidazol de fórmula general (I), donde R = H o alquilo; R' = cadena alquílica interrumpida o no por O, R'' = alquilo o alcoxi; R''' = H o alcoxi substituido o no, caracterizado porque se lleva a cabo mediante la substitución de un grupo nitro en la posición 4 del anillo de piridina de los compuestos de fórmula general (IX) por alcóxido R'O- en presencia de un alcohol R'OH y en el seno de un disolvente polar aprótico. Es útil en el tratamiento o prevención de las úlceras pépticas.
PROCEDIMIENTO PARA PREPARAR N,N,6-TRIMETIL-2-(4-METILFENIL)-IMIDAZO-(1 ,2-A)PIRIDINA-3-ACETAMIDA Y SUS SALES.
Sección de la CIP Química y metalurgia
(01/12/2003). Inventor/es: LABRIOLA, RAFAEL. Clasificación: C07D471/04.
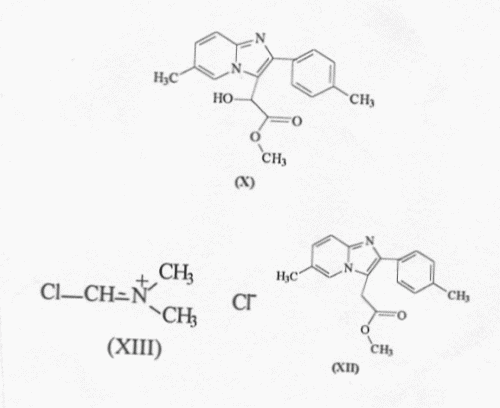
Comprende reducir el hidroxiéster de fórmula (X) haciéndolo reaccionar en DMF sucesivamente con una sal de iminio de fórmula (XIII) formada "in situ" con cloruro de tionilo y dimetilformamida, y reducción subsiguiente con un reductor apropiado para formar el éster de fórmula (XII), que después se hace reaccionar con dimetilamina en un medio solvente polihidroxilado y a una temperatura adecuada.
NUEVA FORMA MEJORADA DE AZITROMICINA MONOHIDRATO DE MENOR HIGROSCOPICIDAD, PROCEDIMIENTO DE PREPARACION Y COMPOSICIONES FARMACEUTICAS QUE LA COMPRENDEN.
Secciones de la CIP Química y metalurgia Necesidades corrientes de la vida
(16/09/2003). Ver ilustración. Inventor/es: BAON PARDO,GABRIEL, FAU DE CASA-JUANA MUÑOZ,MIQUEL, GELPI VINTRO,JOSE MARIA, GONZALEZ HERNANDEZ,PEDRO, LOPEZ GARCIA,JULIA, RIAO RIBOTA,ANA MARIA. Clasificación: C07H17/08, A61P31/04, A61K31/7052.
Nueva forma mejorada de azitromicina monohidrato de menor higroscopicidad, procedimiento de preparación y composiciones farmacéuticas que la comprenden. La forma mejorada de azitromicina monohidrato de menor higroscopicidad se caracteriza por ser más densa y fuerte y por poseer unos valores de: densidad aparente de llenado no inferior a 0,35 g/ml; y densidad aparente golpeada no inferior a 0,50 g/ml. El procedimiento de preparación de una forma mejorada de azitromicina monohidrato, se caracteriza por someter la azitromicina monohidrato higroscópica a un proceso de compactación mediante presión. La invención también se refiere a una composición farmacéutica que comprende una cantidad terapéuticamente efectiva de la forma mejorada de azitromicina monohidrato en asociación con al menos un diluyente inerte adecuado farmacéuticamente aceptable.
NUEVO PROCEDIMIENTO DE OBTENCION DE DERIVADOS DE 2-(2-PIRIDINILMETILSULFINIL)-1H-BENZIMIDAZOL.
Sección de la CIP Química y metalurgia
(01/04/2003). Ver ilustración. Inventor/es: COSME GOMEZ,ANTONIO, FAU DE CASA-JUANA MUÑOZ, MIGUEL, GELPI VINTRO,JOSE MARIA, MOLINA PONCE,ANDRES. Clasificación: C07D401/12.
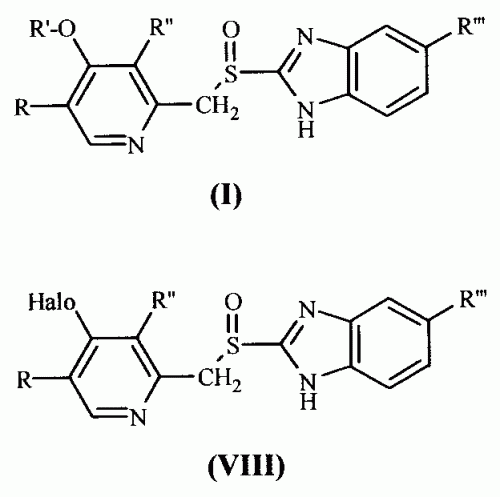
Nuevo procedimiento de obtención de derivados de 2-(2- piridinilmetilsulfinil)-1H-benzimidazol Procedimiento de obtención de un derivado de 2-(2-piridinilmetilsulfinil)-1H- benzimidazol de fórmula general (I), donde R = H; R' = 3- Metoxipropil, R" = metilo; R''' = H, caracterizado porque se lleva a cabo mediante la substitución de un halógeno en la posición 4 del anillo de piridina de los compuestos de fórmula general (VIII) por 3-Metoxipropanol en presencia de una base y en el seno de un disolvente polar aprótico. Es útil en el tratamiento o prevención de las úlceras pépticas.
PROCEDIMIENTO PARA PREPARAR N,N,6-TRIMETIL-2-(4-METILFENIL)-IMIDAZO-(1 ,2-A)-PIRIDINA-3-ACETAMIDA Y SUS SALES.
Sección de la CIP Química y metalurgia
(16/08/2001). Ver ilustración. Inventor/es: LABRIOLA, RAFAEL. Clasificación: C07D471/04.
Procedimiento para preparar n,n,6-trimetil-2-(4-metilfenil)- imidazo-[1,2-a]-piridina-3-acetamida y sus sales. Comprende reducir el hidroxiéster de fórmula (X) haciéndolo reaccionar en DMF sucesivamente con una sal de iminio de fórmula (XIII) formada "in situ" con cloruro de tionilo y dimetilformamida, y reducción subsiguiente con un reductor apropiado para formar el éster de fórmula (XII), que después se hace reaccionar con dimetilamina en un medio solvente polihidroxilado y a una temperatura adecuada.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}