Nuevas estrategias para la secuenciación del genoma.
Procedimiento para determinar la secuencia genómica que comprende las etapas de:
- proporcionar un mapa físico de una muestra de genoma por secuenciación de los extremos de fragmentos defragmentos de clones de cromosomas artificiales mezclados;
- proporcionar un conjunto de lecturas de secuencia de la muestra de genoma;
- generar un cóntigo del mapa físico y las lecturas de secuencia para construir una secuencia genómica.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/NL2010/000003.
Solicitante: KEYGENE N.V..
Nacionalidad solicitante: Países Bajos.
Dirección: P.O. BOX 216 6700 AE WAGENINGEN PAISES BAJOS.
Inventor/es: VAN EIJK,MICHAEL,JOSEPHUS,THERESIA, VAN TUNEN,ADRIANUS JOHANNES, JANSSEN,ANTOINE ANTONIUS ARNOLDUS WILHELMUS.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C12Q1/68 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12Q PROCESOS DE MEDIDA, INVESTIGACION O ANALISIS EN LOS QUE INTERVIENEN ENZIMAS, ÁCIDOS NUCLEICOS O MICROORGANISMOS (ensayos inmunológicos G01N 33/53 ); COMPOSICIONES O PAPELES REACTIVOS PARA ESTE FIN; PROCESOS PARA PREPARAR ESTAS COMPOSICIONES; PROCESOS DE CONTROL SENSIBLES A LAS CONDICIONES DEL MEDIO EN LOS PROCESOS MICROBIOLOGICOS O ENZIMOLOGICOS. › C12Q 1/00 Procesos de medida, investigación o análisis en los que intervienen enzimas, ácidos nucleicos o microorganismos (aparatos de medida, investigación o análisis con medios de medida o detección de las condiciones del medio, p. ej. contadores de colonias, C12M 1/34 ); Composiciones para este fin; Procesos para preparar estas composiciones. › en los que intervienen ácidos nucleicos.
PDF original: ES-2403312_T3.pdf
Fragmento de la descripción:
Nuevas estrategias para la secuenciación del genoma Campo técnico de la invención [0001] La presente invención se refiere a un procedimiento eficaz para la secuenciación del genoma completo de novo. La invención se refiere a la secuenciación de ácidos nucleicos a gran escala y en particular a procedimientos para secuenciar el genoma, o una parte del mismo, de un organismo. La invención se refiere a estrategias mejoradas para determinar la secuencia de, preferiblemente, genomas complejos (es decir grandes) basándose en el uso de tecnologías de secuenciación de alta productividad.
Antecedentes de la invención [0002] El objetivo de muchos proyectos de secuenciación es determinar, por primera vez, la secuencia del genoma completo de un organismo objetivo (secuenciación del genoma borrador de novo) . El tener una secuencia genómica borrador a mano permite la identificación de información genética útil de un organismo, por ejemplo, para identificar el origen de la variedad genética entre especies o individuos de la misma especie. Por lo tanto, hay un deseo general en la materia de llegar a técnicas que permitan la determinación de novo de la secuencia genómica entera de un individuo sea ser humano, animal o planta, con un coste y esfuerzo razonables. Esta búsqueda normalmente se indica como la búsqueda del genoma por 1000 dólares, es decir, determinar la secuencia genómica entera de un individuo por un máximo de 1000 dólares (sin tener en cuenta las fluctuaciones de la moneda) . Sin embargo, en la práctica el genoma por 1000 dólares no se basa necesariamente en la secuenciación genómica de novo y estrategia de ensamblado, sino que se puede basar también en un procedimiento de resecuenciación. En este último caso, el genoma resecuenciado no será ensamblado de novo, sino que su ADN secuenciado será comparado con (cartografiado en) una secuencia genómica de referencia existente para el organismo de interés. Por lo tanto, un procedimiento de resecuenciación es técnicamente menos desafiante y menos costoso. Por motivos de claridad, el foco de la presente invención está en las estrategias de secuenciación genómica de novo, capaces de ser aplicadas a organismos para los que se carece de una secuencia genómica de referencia.
Los esfuerzos actuales están logrando resultados diversos, abundantes y que aumentan rápidamente. No obstante, el objetivo todavía no se ha logrado. Todavía no es económicamente factible secuenciar y ensamblar un genoma completo de una forma directa. Todavía siguen siendo necesarias en la materia estrategias de secuenciación genómica de novo mejoradas.
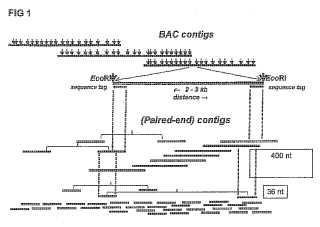
El documento WO03/027311 describe un procedimiento de secuenciación genómica aleatoria (shotgun) de mezcla de matrices de clones (CAPPS) . El procedimiento usa lecturas de secuencia aleatorias de diferentes clones mezclados (BAC) . Basándose en el ensamblado cruzado de las lecturas aleatorias se puede generar un cóntigo de secuencias a partir de una pluralidad de clones y se puede generar un mapa de los clones con respecto a la secuencia. La publicación describe, con más detalle, la generación de una biblioteca de BAC en mezclas multidimensionales, por ejemplo, un formato bidimensional en el que cada mezcla y fila contiene 148 clones de BAC (formato 148 x 148) . Usando CAPPS, las mezclas de BAC se secuencian con una cobertura de 4-5X como media, lo que genera una cobertura de 8-10X por BAC, en el caso del esquema de mezcla bidimensional. Los cóntigos se hacen por BAC por separado basándose en las secuencias que son únicas para el BAC basándose en 45 su aparición en una sola fila y una sola mezcla en el caso de un esquema de mezcla bidimensional. Posteriormente, estos BAC se ensamblan en un cóntigo para el genoma. La publicación demuestra la tecnología basada en 5 BAC solo. La publicación deja sin tocar el problema del procesamiento de datos. Sin embargo, una de las desventajas de esta tecnología es que el uso de fragmentos compartidos aleatoriamente requiere una enorme cantidad de lecturas para cubrir un genoma con un nivel de redundancia de secuencia de 8 a 10 veces, haciendo que este procedimiento 50 sea muy laborioso a gran escala. Además, no da una secuencia basada en el mapa físico de BAC.
El documento US2007/0082358 describe un procedimiento de ensamblado de novo de información de secuencia basado en una biblioteca aislada y amplificada clonalmente de ADN genómico monocatenario para crear la información de secuencia aleatoria del genoma completo combinado con el mapa de restricción óptico del genoma 55 completo usando una enzima de restricción para la creación de un mapa de restricción ordenado.
El documento US2002/0182830 describe un procedimiento de cartografía de cóntigos de BAC por comparación de subsecuencias. El procedimiento se dirige a evitar las dificultades asociadas con secuencias repetitivas y la generación de cóntigos por creación de puentes entre regiones ricas en repeticiones.
La determinación de mapas físicos basados en BAC se puede basar en la secuenciación de bibliotecas de BAC (cartografía física basada en secuencias de clones de BAC) usando por ejemplo, el procedimiento descrito en el documento WO2008/00795 de Keygene indicado también como "perfilado genómico completo" o WPG.
Brevemente, el WPG se refiere a la generación de un mapa físico de al menos parte de un genoma, que comprende las etapas de generar una biblioteca de cromosomas artificiales a partir de una muestra de ADN, mezclar los clones, digerir los clones mezclados con enzimas de restricción, ligar adaptadores que contienen identificador, amplificar los fragmentos de restricción ligados a adaptador que contiene identificador, correlacionar los amplicones con los clones y ordenar los fragmentos para generar un cóntigo para crear así un mapa físico.
El documento WO2008/007951 describe un procedimiento para determinar mapas físicos basados en clones de BAC usando la metodología de WGS.
El Consorcio de secuenciación del genoma del erizo de mar (Science (2006) Vol. 314:941-952)
describe un procedimiento para secuenciar el genoma completo del erizo de mar usando dos estrategias diferentes, CAPSS y WGS, que se combinan para ensamblar el genoma del erizo de mar.
A pesar de todos los desarrollos en la secuenciación de alta productividad, la determinación de secuencias genómicas borrador con gran precisión, todavía se considera caro y laborioso. Sigue siendo necesario complementar los procedimientos que existen actualmente para llegar a procedimientos eficaces y económicos para generar secuencias genómicas borrador. En particular, las tecnologías de secuenciación de alta productividad actuales proporcionan lecturas relativamente cortas (hasta 400 nt) , dando como resultado cóntigos relativamente cortos que son difíciles de ensamblar en cóntigos más largos y requieren una gran demanda de potencia computacional.
Resumen de la invención [0011] Los autores de la presente invención han encontrado que la combinación del perfilado genómico completo basado en clones con la secuenciación (de alta productividad) de fragmentos de muestra de ADN (genómico) 30 usando tecnologías de secuenciación de alta productividad, proporciona una estrategia superior para determinar secuencias genómicas borrador con alta precisión y velocidad. Mediante la generación de cóntigos a partir de las lecturas de secuencia y anclaje de estas lecturas al cóntigo de BAC (o YAC o cualquier otro vector de clonación de inserto grande) obtenido por el perfilado genómico completo, se generan cóntigos de longitud y densidad mayores. Por lo tanto, se obtiene una secuencia genómica borrador que es generada por un número reducido de cóntigos,
aumentado así la calidad de la misma.
Definiciones [0012] Agrupación: con el término "agrupación" se entiende la comparación de dos o más secuencias de nucleótidos basada en la presencia de tramos cortos o largos de nucleótidos idénticos o similares y la agrupación de las secuencias con un determinado nivel mínimo de homología de secuencia basado en la presencia de tramos cortos (o más largos) de secuencias idénticas o similares.
Alineamiento: colocación de múltiples secuencias en una presentación tabular para maximizar la posibilidad
de obtener regiones de identidad de secuencia a lo largo de varias secuencias en el alineamiento, p. ej., mediante la introducción de huecos. Se conocen en la materia varios procedimientos de alineamiento de secuencias de nucleótidos, como se explicará con más detalle a continuación.
AFLP: AFLP se refiere a un procedimiento para la amplificación selectiva de ácidos nucleicos basada en 50 digerir un ácido nucleico con una o más endonucleasas de... [Seguir leyendo]
Reivindicaciones:
1. Procedimiento para determinar la secuencia genómica que comprende las etapas de:
- proporcionar un mapa físico de una muestra de genoma por secuenciación de los extremos de fragmentos de fragmentos de clones de cromosomas artificiales mezclados;
- proporcionar un conjunto de lecturas de secuencia de la muestra de genoma;
- generar un cóntigo del mapa físico y las lecturas de secuencia para construir una secuencia genómica.
2. Procedimiento para determinar una secuencia genómica que comprende las etapas de:
(a) proporcionar una muestra de ADN;
(b) generar un banco de clones de cromosomas artificiales (p. ej., BAC, YAC) en el que cada clon de cromosoma artificial contiene parte de la muestra de ADN;
(c) combinar los clones de cromosomas artificiales en una pluralidad de mezclas, en las que cada clon está presente en más de una mezcla;
(d) proporcionar un conjunto de fragmentos para cada mezcla;
(e) ligar adaptadores a uno o ambos lados de los fragmentos,
(f) determinar la secuencia de al menos parte del adaptador y parte del fragmento; 20 (g) asignar las secuencias de los fragmentos a los correspondientes clones;
(h) construir un cóntigo de clones generando así un mapa físico de la muestra de genoma;
(i) generar lecturas de secuencia de una muestra de ADN;
(j) alinear las lecturas de secuencia y/o cóntigos o andamiajes desde las lecturas de secuencia al cóntigo de
clones para construir así una secuencia genómica/superandamiaje. 25
3. Procedimiento de acuerdo con la reivindicación 2, en el que al menos un adaptador contiene un identificador específico de mezcla o una sección de identificador degenerado, respectivamente, para proporcionar fragmentos ligados a adaptador que contienen identificador.
4. Procedimiento de acuerdo con las reivindicaciones 2 ó 3, en el que los fragmentos ligados a un adaptador se amplifican usando
- un cebador que amplifica al menos el identificador y parte del fragmento; o
- un cebador que contiene una sección que es complementaria de la sección degenerada en el adaptador e 35 introduce un identificador en el fragmento amplificado; o
- un cebador que es complementario de al menos parte del adaptador y proporciona un identificador en el fragmento ligado a adaptador amplificado.
5. Procedimiento de acuerdo con las reivindicaciones 2-4, en el que los fragmentos para una mezcla se 40 generan por fragmentación aleatoria de las mezclas y/o fragmentación con enzimas de restricción de las mezclas.
6. Procedimiento de acuerdo con las reivindicaciones 2-5, en el que las lecturas de secuencia se obtienen de la muestra de ADN fragmentada y/o de uno o más clones de cromosomas artificiales de la muestra de ADN.
7. Procedimiento de acuerdo con las reivindicaciones 2-6, en el que las lecturas de secuencia se obtienen de la muestra de ADN fragmentada aleatoriamente y/o de uno o más clones de cromosomas artificiales de la muestra de ADN.
8. Procedimiento de acuerdo con las reivindicaciones 2-6, en el que las lecturas de secuencia se obtienen de fragmentos de restricción que se han obtenido por fragmentación con enzimas de restricción de la muestra de ADN y/o de uno o más clones de cromosomas artificiales de la muestra de ADN.
9. Procedimiento de acuerdo con la reivindicación 8, en el que los fragmentos de restricción son 55 fragmentos de restricción ligados a adaptador.
10. Procedimiento de acuerdo con la reivindicación 9, en el que los fragmentos de restricción ligados a adaptador se amplifican de forma selectiva o no selectiva.
11. Procedimiento de acuerdo con cualquiera de las reivindicaciones anteriores, en el que la secuenciación se lleva a cabo mediante secuenciación de alta productividad.
12. Procedimiento de acuerdo con la reivindicación 11, en el que la secuenciación de alta productividad se 5 realiza sobre un soporte sólido.
13. Procedimiento de acuerdo con la reivindicación 11 ó 12, en el que la secuenciación de alta productividad se basa en secuenciación por síntesis.
14. Procedimiento de acuerdo con la reivindicación 11 ó 12, en el que la secuenciación se basa en pirosecuenciación.
Patentes similares o relacionadas:
Método para analizar ácido nucleico molde, método para analizar sustancia objetivo, kit de análisis para ácido nucleico molde o sustancia objetivo y analizador para ácido nucleico molde o sustancia objetivo, del 29 de Julio de 2020, de Kabushiki Kaisha DNAFORM: Un método para analizar un ácido nucleico molde, que comprende las etapas de: fraccionar una muestra que comprende un ácido nucleico molde […]
MÉTODOS PARA EL DIAGNÓSTICO DE ENFERMOS ATÓPICOS SENSIBLES A COMPONENTES ALERGÉNICOS DEL POLEN DE OLEA EUROPAEA (OLIVO), del 23 de Julio de 2020, de SERVICIO ANDALUZ DE SALUD: Biomarcadores y método para el diagnostico, estratificación, seguimiento y pronostico de la evolución de la enfermedad alérgica a polen del olivo, kit […]
Detección de interacciones proteína a proteína, del 15 de Julio de 2020, de THE GOVERNING COUNCIL OF THE UNIVERSITY OF TORONTO: Un método para medir cuantitativamente la fuerza y la afinidad de una interacción entre una primera proteína de membrana o parte de la misma y una […]
Secuenciación dirigida y filtrado de UID, del 15 de Julio de 2020, de F. HOFFMANN-LA ROCHE AG: Un procedimiento para generar una biblioteca de polinucleótidos que comprende: (a) generar una primera secuencia del complemento (CS) de un polinucleótido diana a partir […]
Métodos para la recopilación, estabilización y conservación de muestras, del 8 de Julio de 2020, de Drawbridge Health, Inc: Un método para estabilizar uno o más componentes biológicos de una muestra biológica de un sujeto, comprendiendo el método obtener un […]
Evento de maíz DP-004114-3 y métodos para la detección del mismo, del 1 de Julio de 2020, de PIONEER HI-BRED INTERNATIONAL, INC.: Un amplicón que consiste en la secuencia de ácido nucleico de la SEQ ID NO: 32 o el complemento de longitud completa del mismo.
Aislamiento de ácidos nucleicos, del 24 de Junio de 2020, de REVOLUGEN LIMITED: Un método de aislamiento de ácidos nucleicos que comprenden ADN de material biológico, comprendiendo el método las etapas que consisten en: (i) efectuar un lisado […]
Composiciones para modular la expresión de SOD-1, del 24 de Junio de 2020, de Biogen MA Inc: Un compuesto antisentido según la siguiente fórmula: mCes Aeo Ges Geo Aes Tds Ads mCds Ads Tds Tds Tds mCds Tds Ads mCeo Aes Geo mCes Te (secuencia […]