FORMULACIONES OPIOIDES ADMINISTRABLES ORALMENTE OBTENIDAS MEDIANTE EXTRUSION POR FUSION.
Forma de dosificación oral, analgésica, de liberación sostenida,
que comprende una masa extruída en fusión, farmacéutica, que incluye un analgésico opioide en forma de una combinación antagonista mu dispersada en una matriz, en la que la matriz comprende por lo menos un material hidrófobo, y un segundo retardante seleccionado de entre ceras, alcoholes grasos, ácidos grasos y mezclas de los mismos
Tipo: Patente Europea. Resumen de patente/invención. Número de Solicitud: E03015267.
Solicitante: EURO-CELTIQUE S.A..
Nacionalidad solicitante: Luxemburgo.
Dirección: 122 BOULEVARD DE LA PETRUSSE,2330 LUXEMBOURG.
Inventor/es: OSHLACK, BENJAMIN, CHASIN, MARK, HUANG, HUA-PIN, SACKLER, DAVID.
Fecha de Publicación: .
Fecha Solicitud PCT: 3 de Noviembre de 1995.
Fecha Concesión Europea: 23 de Diciembre de 2009.
Clasificación Internacional de Patentes:
- A61K9/16H4
- A61K9/16H6B
- A61K9/16H6F
- A61K9/16P4
- A61K9/20H4B
- A61K9/20H6B
- A61K9/20K2
- A61K9/20K2B
- A61K9/20P
Clasificación PCT:
- A61K9/16 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 9/00 Preparaciones medicinales caracterizadas por un aspecto particular. › Aglomerados; Granulados; Microbolitas.
- A61K9/20 A61K 9/00 […] › Píldoras, pastillas o comprimidos.
Clasificación antigua:
Países PCT: Austria, Bélgica, Suiza, Alemania, Dinamarca, España, Francia, Reino Unido, Grecia, Italia, Liechtensein, Luxemburgo, Países Bajos, Suecia, Mónaco, Portugal, Irlanda.
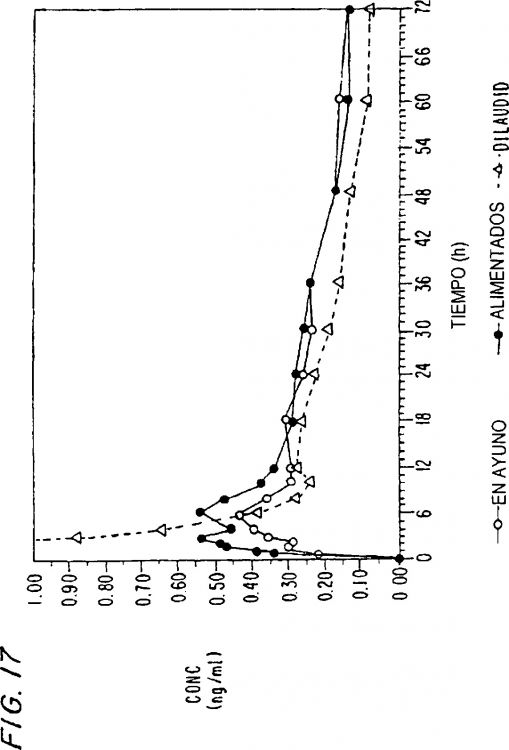
Fragmento de la descripción:
Formulaciones opioides administrables oralmente obtenidas mediante extrusión por fusión.
Antecedentes de la invención
La presente invención se refiere a la utilización de la tecnología de extrusión por fusión en la producción de formulaciones farmacéuticas biodisponibles de matrices de liberación sostenida. Anteriormente, la extrusión por fusión se ha utilizado en la producción de formulaciones de liberación inmediata.
En la técnica farmacéutica se conoce la preparación de composiciones que proporcionan una liberación controlada de sustancias farmacológicamente activas contenidas en las composiciones después de su administración oral a humanos y animales. Dichas composiciones de liberación lenta se utilizan para retrasar la absorción de un medicamento hasta que ha alcanzado ciertas partes del tracto alimentario. Dicha liberación sostenida de un medicamento en el tracto alimentario mantiene además una concentración deseada de dicho medicamento en la corriente sanguínea durante más tiempo que el que se produciría si se administrasen formas de dosificación de liberación rápida convencionales.
Se han sugerido diferentes métodos de preparación de formas de dosificación farmacéuticas de liberación controlada. Por ejemplo, se han propuesto técnicas de compresión directa, técnicas de granulación por vía húmeda, técnicas de encapsulación y similares para suministrar ingredientes farmacéuticamente activos al tracto alimentario durante periodos de tiempo prolongados.
Adicionalmente, en la técnica se conocen varios tipos de formulaciones de liberación sostenida, incluyendo especialmente pellets recubiertos, comprimidos recubiertos y cápsulas en los que la liberación lenta del medicamento activo se produce a través de la descomposición selectiva del recubrimiento de la preparación o a través de la formación de compuestos con una matriz especial para influir en la liberación de un fármaco. Algunas formulaciones de liberación sostenida proporcionan una liberación secuencial relacionada de una dosis individual de un compuesto activo en periodos predeterminados después de la administración.
El propósito de todas las preparaciones de liberación sostenida es proporcionar un periodo más largo de respuesta farmacológica después de la administración del fármaco y se experimenta normalmente después de la administración de las formas de dosificación de liberación rápida. Dichos periodos más largos de respuesta proporcionan muchos beneficios terapéuticos inherentes que no se consiguen con preparaciones correspondientes de acción corta y liberación inmediata. Esto se cumple especialmente en el tratamiento de pacientes con cáncer u otros pacientes que necesitan un tratamiento para aliviar un dolor de moderado a severo, en los que los niveles en sangre de un medicamento analgésico opioide se debe mantener en un nivel terapéuticamente eficaz para proporcionar el alivio del dolor. A no ser que la terapia convencional de fármacos de acción rápida se administre cuidadosamente a intervalos frecuentes para mantener unos niveles en sangre eficaces del fármaco en estado de equilibrio, en el nivel en sangre del fármaco activo se producen picos y depresiones debido a la absorción rápida, la excreción sistémica del compuesto y a través de la inactivación metabólica, generando de este modo problemas especiales en el mantenimiento de la eficacia analgésica.
Las enseñanzas de la técnica anterior sobre la preparación y utilización de composiciones que proporcionan la liberación sostenida de un compuesto activo a partir de un vehículo se refieren básicamente a la liberación de la sustancia activa en el fluido fisiológico del tracto alimentario. No obstante, en general se reconoce que la mera presencia de una sustancia activa en los fluidos gastrointestinales no garantiza, por sí misma, la biodisponibilidad.
Para que sea absorbida, la sustancia farmacológica activa debe estar en solución. El tiempo requerido para una proporción determinada de una sustancia activa a partir de una forma de dosificación unitaria se determina como la proporción de la cantidad de sustancia farmacológica activa liberada a partir de una forma de dosificación unitaria durante una base de tiempos especificada mediante un método de prueba realizado en condiciones normalizadas. Los fluidos fisiológicos del tracto gastrointestinal son los medios para determinar el tiempo de disolución. El estado actual de la técnica reconoce muchos procedimientos de prueba satisfactorios para medir el tiempo de disolución para composiciones farmacéuticas, y estos procedimientos de prueba se describen en compendios oficiales en todo el mundo.
Aunque existen muchos factores diferentes que influyen en la disolución de una sustancia farmacológica desde su vehículo, el tiempo de disolución determinado para una sustancia farmacológicamente activa a partir de la composición específica es relativamente constante y reproducible. Entre los diferentes factores que afectan al tiempo de disolución se encuentran el área de la superficie de la sustancia farmacológica presentada al medio disolvente de la disolución, el pH de la disolución, la solubilidad de la sustancia en el medio disolvente específico, y las fuerzas impulsoras de la concentración de saturación de materiales disueltos en el medio disolvente. De este modo, la concentración de la disolución de una sustancia farmacológica activa se modifica dinámicamente en su estado de equilibrio a medida que los componentes se eliminan del medio de la disolución debido a la absorción a través del lugar tisular. En condiciones fisiológicas, el nivel de saturación de los materiales disueltos se restablece a partir de la reserva de la forma de dosificación para mantener una concentración de la disolución relativamente uniforme y constante en el medio disolvente proporcionando una absorción en estado de equilibrio.
En el transporte a través de un lugar de absorción tisular del tracto gastrointestinal influyen las fuerzas de equilibrio osmótico de Donnan a ambos lados de la membrana ya que la dirección de la fuerza impulsora es la diferencia entre las concentraciones de la sustancia activa a cada lado de la membrana, es decir, la cantidad disuelta en los fluidos gastrointestinales y la cantidad presente en la sangre. Como los niveles de sangre están siendo modificados constantemente por la dilución, los cambios circulatorios, el almacenamiento en el tejido, la conversión metabólica y la excreción sistémica, el flujo de materiales activos se dirige desde el tracto gastrointestinal hacia la corriente sanguínea.
A pesar de los diversos factores que influyen tanto en la disolución como en la absorción de una sustancia farmacológica, se ha establecido una fuerte correlación entre el tiempo de disolución in vitro determinado para una forma de dosificación y la biodisponibilidad (in vivo). El tiempo de disolución y la biodisponibilidad determinados para una composición son dos de las características fundamentales más significativas a considerar cuando se evalúan composiciones de liberación sostenida.
También se han sugerido técnicas de granulación por fusión para proporcionar formulaciones de liberación controlada. En general, la granulación por fusión implica el procesado mecánico de un ingrediente activo en forma de partículas con uno o más aglutinantes adecuados y/o excipientes farmacéuticamente aceptables en un mezclador hasta que uno o más de los aglutinantes se funde y se adhiere a la superficie de las partículas, constituyendo finalmente gránulos.
La patente U.S. n° 4.957.681 (Klimesch et al.) da a conocer un proceso continuo para preparar mezclas farmacéuticas que tienen por lo menos dos componentes los cuales se dosifican continuamente. El proceso incluye la dosificación continua de los componentes individuales de la mezcla farmacéutica a una velocidad de por lo menos 50 g/h en balanzas de medición diferencial electrónica que tienen una precisión de medición de por lo menos
Reivindicaciones:
1. Forma de dosificación oral, analgésica, de liberación sostenida, que comprende una masa extruída en fusión, farmacéutica, que incluye un analgésico opioide en forma de una combinación antagonista mu dispersada en una matriz, en la que la matriz comprende por lo menos un material hidrófobo, y un segundo retardante seleccionado de entre ceras, alcoholes grasos, ácidos grasos y mezclas de los mismos.
2. Forma de dosificación de la reivindicación 1, en la que la masa extruída tiene la forma de múltiples partículas.
3. Forma de dosificación de la reivindicación 2, en la que las múltiples partículas se obtienen cortando una hebra de masa extruída con un diámetro de entre 0,1 y 5 mm.
4. Forma de dosificación de la reivindicación 2 ó 3, en la que las múltiples partículas tienen una longitud en un intervalo de entre 0,1 y 12 mm y presentan un diámetro de entre 0,1 y 5 mm.
5. Forma de dosificación de una cualquiera de las reivindicaciones 1 a 4, en la que el por lo menos un material hidrófobo se selecciona del grupo consistente en alquilcelulosas, polímeros y copolímeros de ácidos acrílico y metacrílico, goma laca, ceína, aceite de ricino hidrogenado o aceite vegetal hidrogenado, o mezclas de los mismos.
6. Forma de dosificación de una cualquiera de las reivindicaciones 1 a 4, en la que el por lo menos un material hidrófobo es etilcelulosa.
7. Forma de dosificación de una cualquiera de las reivindicaciones 1 a 6, en la que la matriz comprende otros materiales seleccionados del grupo de diluyentes, lubricantes, aglutinantes, medios granuladores, colorantes, aromatizantes y deslizantes que son convencionales en la técnica farmacéutica.
8. Forma de dosificación de una cualquiera de las reivindicaciones 1 a 7, en la que la forma de dosificación comprende la masa extruída en forma de múltiples partículas que están recubiertas con un recubrimiento de liberación sostenida.
9. Forma de dosificación de la reivindicación 8, en la que el recubrimiento comprende un material hidrófobo.
10. Forma de dosificación de una cualquiera de las reivindicaciones 1 a 9, en la que la forma de dosificación es una cápsula.
11. Forma de dosificación de una cualquiera de las reivindicaciones 1 a 10, que proporciona un efecto analgésico durante por lo menos 8 horas después de su administración a un paciente humano.
12. Forma de dosificación de una cualquiera de las reivindicaciones 1 a 10, que proporciona un efecto analgésico durante por lo menos 12 horas después de su administración a un paciente humano.
13. Forma de dosificación de una cualquiera de las reivindicaciones 1 a 10, que proporciona un efecto analgésico durante aproximadamente 24 horas después de su administración a un paciente humano.
Patentes similares o relacionadas:
COMPOSICIÓN FARMACÉUTICA CON INGREDIENTE ACTIVO ATORVASTATINA, del 6 de Febrero de 2012, de ZENTIVA, K.S: Composición farmacéutica que comprende la sustancia activa atorvastatina, diseñada para el tratamiento y prevención de enfermedades cardiovasculares, […]
COMPOSICIÓN FARMACÉUTICA ORAL ESTABLE QUE CONTIENE AGONISTAS DE RECEPTOR DE HORMONAS TIROIDEAS, del 2 de Diciembre de 2011, de KARO BIO AB BRISTOL-MYERS SQUIBB COMPANY: Una composición farmacéutica adecuada para administración oral, que comprende: (i) un compuesto de Fórmula I: Fórmula I en la que: Z es H o un grupo alternativo capaz de […]
FORMULACIONES SÓLIDAS DE OSPEMIFENO, del 19 de Septiembre de 2011, de HORMOS MEDICAL LTD.: Una formulación sólida de un fármaco, que comprende granulados que contienen 30 a 90 mg de ospemifeno o una de sus sales farmacéuticamente aceptables en combinación con una […]
PREPARACIÓN FARMACÉUTICA QUE CONTIENE GABAPENTINA, del 5 de Agosto de 2011, de ZAMBON S.P.A.: Un granulado de gabapentina obtenido por granulación en estado fundido de gabapentina con PEG, que tiene un punto de fusión comprendido entre […]
COMPOSICIONES FARMACÉUTICAS QUE CONTIENEN IRBESARTÁN Y UN DIURÉTICO, del 21 de Julio de 2011, de SANOFI-AVENTIS: Una composición farmacéutica que comprende, en peso: - 50% de irbesartán; - 8,33% de hidroclorotiazida y 4,72% de lactosa hidratada o 4,17% de hidroclorotiazida […]
COMPOSICIONES EN COMPRIMIDOS QUE CONTIENEN ATAZANAVIR, del 22 de Junio de 2011, de BRISTOL-MYERS SQUIBB COMPANY: Un comprimido que comprende ritonavir y gránulos que contienen sulfato de atazanavir y un lubricante intragranular, teniendo dicho gránulo […]
COMPOSICIONES EN COMPRIMIDOS QUE CONTIENEN ATAZANAVIR, del 3 de Junio de 2011, de BRISTOL-MYERS SQUIBB COMPANY: Un comprimido que comprende raltegravir y gránulos que contienen sulfato de atazanavir y un lubricante intragranular, teniendo dichos gránulos una sección […]
COMPOSICIONES EN COMPRIMIDOS QUE CONTIENEN ATAZANAVIR, del 26 de Mayo de 2011, de BRISTOL-MYERS SQUIBB COMPANY: Un comprimido que comprende sulfato de abacavir y gránulos que contienen sulfato de atazanavir y un lubricante intragranular, teniendo dichos gránulos una sección interior […]