USO DEL ENANTIÓMERO (1S,2R) DEL MILNACIPRÁN PARA LA PREPARACIÓN DE UN MEDICAMENTO.
Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida),
o una de sus sales farmacéuticamente aceptables, para su utilización como medicamento destinado a prevenir o a tratar la depresión, los estados depresivos, la fibromialgia, el síndrome de fatiga crónica, el dolor, en pacientes que presentan unos antecedentes cardiovasculares y/o que padecen trastornos cardiovasculares, administrado a una dosis comprendida entre 0,01 mg y 10 mg/kg de peso corporal por día en una o varias tomas
Tipo: Patente Europea. Resumen de patente/invención. Número de Solicitud: E07123564.
Solicitante: PIERRE FABRE MEDICAMENT.
Nacionalidad solicitante: Francia.
Dirección: 45, PLACE ABEL GANCE 92100 BOULOGNE-BILLANCOURT FRANCIA.
Inventor/es: DEREGNAUCOURT,JEAN, GROSSE,RICHARD.
Fecha de Publicación: .
Fecha Solicitud PCT: 16 de Febrero de 2004.
Clasificación Internacional de Patentes:
- A61K31/135 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › que tienen ciclos aromáticos, p. ej. metadona.
- A61K31/165 A61K 31/00 […] › teniendo ciclos aromáticos, p. ej. colchicina, atenolol, progabide.
- A61K31/167 A61K 31/00 […] › teniendo el átomo de nitrogeno de un grupo carboxiamida unido directamente al ciclo aromático, p. ej. lidocaina, paracetamol.
- A61K31/195 A61K 31/00 […] › que tienen un grupo amino.
- A61K31/4015 A61K 31/00 […] › teniendo grupos oxo unidos directamente al heterociclo, p. ej. piracetam, etosuximida.
Clasificación PCT:
- A61K31/135 A61K 31/00 […] › que tienen ciclos aromáticos, p. ej. metadona.
- A61K31/165 A61K 31/00 […] › teniendo ciclos aromáticos, p. ej. colchicina, atenolol, progabide.
Países PCT: Austria, Bélgica, Suiza, Alemania, Dinamarca, España, Francia, Reino Unido, Grecia, Italia, Liechtensein, Luxemburgo, Países Bajos, Suecia, Mónaco, Portugal, Irlanda, Eslovenia, Finlandia, Rumania, Chipre, Lituania, Letonia, Ex República Yugoslava de Macedonia, Albania.
PDF original: ES-2359441_T3.pdf



Fragmento de la descripción:
40
45
50
La presente invención se refiere al enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1fenilciclopropanocarboxamida), o de una de sus sales farmacéuticamente aceptables, para su utilización como medicamento destinado a prevenir o a tratar la depresión, los estados depresivos, la fibromialgia, el síndrome de fatiga crónica, el dolor, en pacientes que presentan unos antecedentes cardiovasculares y/o que padecen trastornos cardiovasculares, administrado a una dosis comprendida entre 0,01 mg y 10 mg/kg de peso corporal por día en una
o varias tomas.
El Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), molécula sintetizada en el Centro de Investigación PIERRE FABRE MEDICAMENT (Castres, Francia), también denominado TN-912, Dalciprán, Milnaciprán, Midalciprán o Midaliprán, se conoce como inhibidor doble de la recaptura de la serotonina (5-HT) y de la noradrenalina (NA). El Milnaciprán y su procedimiento de preparación se describen en la patente US nº 4.478.836. Otras informaciones relativas al Milnaciprán se pueden encontrar en la duodécima edición del índice Merck, con la entrada nº 6281.
Los inhibidores dobles de la recaptura de la serotonina y de la noradrenalina corresponden a una clase bien conocida de agentes antidepresivos que inhiben de manera selectiva la recaptura al mismo tiempo de la serotonina y de la noradrenalina. A título de ejemplo, la venlafaxina y la duloxetina son asimismo unos inhibidores dobles de la serotonina y de la noradrenalina. Unos estudios han mostrado que el ratio entre la inhibición de la recaptura de la noradrenalina y la inhibición de la serotonina mediante el Milnaciprán es de aproximadamente 2:1 (Moret et al., 1985 Neuropharmacology 24(12): 1211-1219; Palmier et al., 1989, Eur. J. Clin. Pharmacol 37: 235-238).
La patente US nº 4.478.836 describe el uso del Milnaciprán para el tratamiento de patologías del sistema nervioso central, en particular de la depresión. La solicitud de patente WO 01/26623 describe el uso del Milnaciprán en asociación con fenilalanina y tirosina en unas indicaciones tales como el tratamiento de la fatiga, de los síndromes asociados con el dolor, del síndrome de la fatiga crónica, de la fibromialgia, del síndrome del intestino irritable. La solicitud de patente WO 01/62236 describe una composición que comprende Milnaciprán en asociación con uno o varios agentes anti-muscarínicos en un gran número de indicaciones incluyendo la depresión. La solicitud WO 97/35574 describe una composición farmacéutica que contiene Milnaciprán e idazoxano como producto de combinación para un uso simultáneo, separado o espaciado en el tiempo para tratar la depresión y sus diferentes formas, así como las patologías en las que se usan los antidepresivos. El Milnaciprán se usa asimismo en una indicación de tratamiento de la incontinencia urinaria (FR 2 759 290).
La molécula de Milnaciprán posee dos carbonos asimétricos que llevan a dos configuraciones espaciales diferentes (1S,2R) y (1R,2S). Siendo estas configuraciones espaciales no superponibles, la molécula de Milnaciprán presenta por lo tanto una isomería óptica.
El clorhidrato de Milnaciprán existe así en forma de dos enantiómeros ópticamente activos: el enantiómero dextrógiro o también clorhidrato de Z-(1S,2R)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida y el enantiómero levógiro clorhidrato de Z-(1R, 2S)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida. El Milnaciprán en su forma de clorhidrato (también denominado F2207) está actualmente comercializado (IXEL, PIERRE FABRE MEDICAMENT, Francia) en forma de mezcla racémica como medicamento antidepresivo serotoninérgico-noradrenérgico. F2695 y F2696 designan, respectivamente, los enantiómeros (1S,2R) (dextrógiro) y (1R,2S) (levógiro) del clorhidrato de Milnaciprán (F2207):
**(Ver fórmula)**
Estos dos enantiómeros pueden estar separados o aislados según unos procedimientos descritos en la bibliografía (Bonnaud et al., 1985, Journal of Chromatography, Vol. 318: 398-403; Shuto et al., Tetrahedron letters, 1996 Vol. 37:641-644; Grard et al., 2000, Electrophoresis 2000 21: 3028-3034; Doyle y Hu, 2001, Advanced Synthesis and Catalysis, Vol. 343: 299-302).
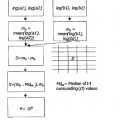
Los inventores han realizado ahora un estudio farmacocinético en el ser humano del racemato y de los dos enantiómeros del Milnaciprán que usa unos métodos de dosificación de enantio-selectividad. Así, han demostrado la ausencia de racemización de los enantiómeros in vivo.
Por otra parte, aunque el racemato haya sido resuelto, no se ha realizado ningún análisis de las propiedades farmacológicas y toxicológicas de los dos enantiómeros, que usa los métodos modernos actualmente a disposición tales como las mediciones cardiovasculares por telemetría, o los análisis de predictividad farmaco-toxico-genómica in vitro.
Los antidepresivos, como cualquier principio activo, pueden generar unos efectos indeseables o ciertas toxicidades que resultan esencialmente de las propiedades farmacológicas de estos medicamentos, pero asimismo de la posología, de la variabilidad individual del paciente (polimorfismo genético, insuficiencia orgánica, sexo, edad) o de interacciones medicamentosas. Así, los antidepresivos representan la tercera clase de productos responsables de intoxicación, después de los hipnóticos y de los tranquilizantes (Nores et al., 1987 Thérapie 42: 555-558). El riesgo de sobredosis con unos antidepresivos es grave puesto que puede llevar a la muerte. Entre las causas de intoxicaciones agudas mediante los antidepresivos, conviene citar la ingesta involuntaria por los niños (considerando que ciertos antidepresivos se usan para el tratamiento de la enuresis), el intento de suicidio, la sobredosis involuntaria por el médico, la co-medicación asociada en la persona mayor, las modificaciones fisiológicas y farmacocinéticas relacionadas con la edad (insuficiencia cardiaca, insuficiencia hepática y/o renal, etc.), un metabolismo ralentizado de origen genético o medicamentoso (inhibición enzimática). Después de los niños, la persona mayor constituye por lo tanto la segunda población de riesgo entre los pacientes tratados. Estas personas tienen unas concentraciones plasmáticas más elevadas, relacionadas con una aclaración renal y/o hepática disminuida, y los riesgos de intoxicación son más graves (Meadoer-Woodruff et al., 1988 J. Clim. Psychopharmacol.
8: 28-32).
Los efectos secundarios indeseables, generalmente benignos, observados durante el tratamiento por el Milnaciprán se observan sobretodo durante la primera, incluso las dos primeras semanas del tratamiento, y se difuminan después, paralelamente con la mejora del episodio depresivo. Los eventos indeseables relatados más comúnmente en monoterapia, o durante una asociación con otros psicótropos son unos vértigos, una hipersudoración, la ansiedad, unos sofocos, y la disuria. Ciertos efectos indeseables relatados menos comúnmente son unas nauseas, unos vómitos, una sequedad bucal, un estreñimiento, unos temblores, unas palpitaciones, una agitación, unas erupciones cutáneas. Por otra parte, se conoce en los pacientes que presentan unos antecedentes cardiovasculares
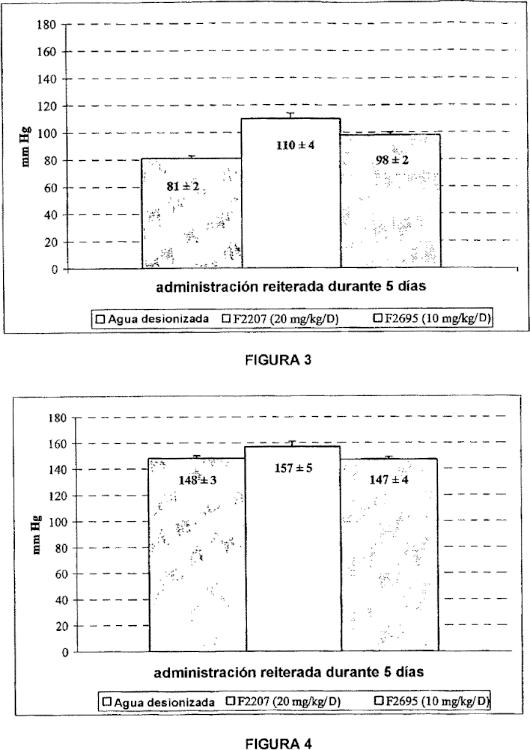
o que reciben simultáneamente un tratamiento con diana cardiaca, que la incidencia de los efectos indeseables de naturaleza cardiovascular (hipertensión, hipotensión, hipotensión ortostática, palpitaciones) puede estar aumentada por el Milnaciprán. En los pacientes hipertensos o que padecen cardiopatías, se recomienda así reforzar la vigilancia clínica porque el Milnaciprán en forma de mezcla racémica es susceptible de aumentar la frecuencia cardiaca. Así, durante raros casos de sobredosis observados con el Milnaciprán (con dosis de 800 mg a 1 g) en monoterapia, los principales síntomas observados son los vómitos, los trastornos respiratorios y una taquicardia (Dictionnaire Vidal, 78ª edición, 2002). Otro efecto indeseable excepcionalmente inducido por el Milnaciprán es un aumento elevado de las transaminasas, que puede traducir una cierta toxicidad hepática.
De hecho, las poblaciones de riesgo susceptibles de desarrollar un cierto número de manifestaciones clínicas indeseables durante o después de un tratamiento por el Milnaciprán, son los insuficientes hepáticos y/o renales, los pacientes que reciben un tratamiento que induce unas toxicidades orgánicas y/o tisulares, en particular unas toxicidades hepáticas y/o renales, los pacientes que reciben un tratamiento... [Seguir leyendo]
Reivindicaciones:
REIVINDICAIONES
1. Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), o una de sus sales farmacéuticamente aceptables, para su utilización como medicamento destinado a prevenir o a tratar la depresión, los estados depresivos, la fibromialgia, el síndrome de fatiga crónica, el dolor, en pacientes que presentan unos antecedentes cardiovasculares y/o que padecen trastornos cardiovasculares, administrado a una dosis comprendida entre 0,01 mg y 10 mg/kg de peso corporal por día en una o varias tomas.
2. Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), o una de sus sales farmacéuticamente aceptables según la reivindicación 1, caracterizado porque la dosis administrada está comprendida entre 0,05 mg y 5 mg/kg de peso corporal por día en una o varias tomas.
3. Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), o una de sus sales farmacéuticamente aceptables según la reivindicación 1 ó 2, caracterizado porque la dosis administrada está comprendida entre 0,1 mg y 1 mg/kg de peso corporal por día en una o varias tomas.
4. Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), o una de sus sales farmacéuticamente aceptables según cualquiera de las reivindicaciones 1 a 3, caracterizado porque los trastornos cardiovasculares corresponden a un aumento de la presión arterial y/o a un aumento de la frecuencia cardiaca.
5. Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), o una de sus sales farmacéuticamente aceptables según la reivindicación 4, caracterizado porque el aumento de la presión arterial corresponde a un aumento de la presión arterial diastólica.
6. Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), o una de sus sales farmacéuticamente aceptables según cualquiera de las reivindicaciones 1 a 5, limitando además al mismo tiempo los riesgos de toxicidad orgánica y/o tisular.
7. Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), o una de sus sales farmacéuticamente aceptables según cualquiera de las reivindicaciones 1 a 6, caracterizado porque dicho enantiómero (1S,2R) del Milnaciprán es el clorhidrato de Z-(1S,2R)-2-(aminometil)-N,N-dietil-1fenilciclopropanocarboxamida (F2695).
8. Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), o una de sus sales farmacéuticamente aceptables según cualquiera de las reivindicaciones 1 a 7, para el tratamiento o la prevención de la depresión profunda, de la depresión resistente, de la depresión de la persona mayor, de la depresión psicótica, de la depresión inducida por unos tratamientos con interferón, del estado depresivo, del síndrome maniaco-depresivo, de los episodios depresivos estacionales, de los episodios depresivos relacionados con una condición médica general o de los episodios depresivos relacionados con unas sustancias que actúan sobre el humor.
9. Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), o una de sus sales farmacéuticamente aceptables según cualquiera de las reivindicaciones 1 a 7, para el tratamiento o la prevención del dolor crónico.
10. Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), o una de sus sales farmacéuticamente aceptables según cualquiera de las reivindicaciones 1 a 9, caracterizado porque los antecedentes cardiovasculares y/o trastornos cardiovasculares se seleccionan de entre el infarto de miocardio, los trastornos del ritmo cardiaco (taquicardia, bradicardia, palpitaciones), los trastornos de la presión arterial (pacientes hipo-o hipertensos) y las cardiopatías.
11. Enantiómero (1S,2R) del Milnaciprán (Z(±)-2-(aminometil)-N,N-dietil-1-fenilciclopropanocarboxamida), o una de sus sales farmacéuticamente aceptables según cualquiera de las reivindicaciones 1 a 10, caracterizado porque el medicamento contiene:
a) dicho enantiómero (1S,2R) del Milnaciprán o una de sus sales farmacéuticamente aceptables, y
b) por lo menos un compuesto activo seleccionado de entre los psicótropos, en particular los antidepresivos, y los agentes anti-muscarínicos,
como productos de combinación para un uso simultáneo, separado o espaciado en el tiempo para el tratamiento o la prevención de la depresión, en particular la depresión profunda, la depresión resistente, la depresión de la persona mayor, la depresión psicótica, la depresión inducida por unos tratamientos con interferón, el estado depresivo, el síndrome maniaco-depresivo, los episodios depresivos estacionales, los episodios depresivos relacionados con una condición médica general, o los episodios depresivos relacionados con unas sustancias que actúan sobre el humor.
Patentes similares o relacionadas:
Uso de (1R,2R)-3-(3-dimetilamino-1-etil-2-metil-propil)-fenol para tratar el dolor inflamatorio, del 22 de Julio de 2020, de GRUNENTHAL GMBH: (1R, 2R)-3-(3-Dimetilamino-1-etil-2-metil-propil)-fenol para uso en el tratamiento del dolor inflamatorio.
Régimen de dosificación para un agonista del receptor S1P, del 3 de Junio de 2020, de NOVARTIS AG: Uso de un modulador o agonista del receptor S1P en la fabricación de un medicamento para el tratamiento de una enfermedad autoinmune, mediante […]
Métodos de administración de terapia con pirfenidona, del 13 de Mayo de 2020, de Intermune, Inc: Pirfenidona para su uso en el tratamiento de un paciente que necesita terapia con pirfenidona, caracterizada por que el tratamiento comprende reducir la dosis de […]
Composiciones congeladas fluidas que comprenden un agente terapéutico, del 8 de Abril de 2020, de Tavakoli, Zahra: Una composición fluida congelada que comprende un agente terapéutico y al menos un agente aromatizante para su uso en terapia, donde dicha composición […]
Sistema terapéutico transdérmico para la administración de principios activos hidrosolubles, del 18 de Marzo de 2020, de LTS LOHMANN THERAPIE-SYSTEME AG: Sistema terapéutico transdérmico para la administración controlada de un principio activo farmacéutico soluble en agua a partir de una fase acuosa que comprende una […]
COMBINACIÓN FARMACÉUTICA SINÉRGICA QUE COMPRENDE TRAMADOL CLORHIDRATO Y PREGABALINA, Y SU USO PARA EL TRATAMIENTO DEL DOLOR NEUROPÁTICO, del 5 de Marzo de 2020, de GRUNENTHAL GMBH: Combinación farmacéutica sinérgica que contiene tramadol clorhidrato y pregabalina en una razón p/p entre 1 :1,5 a 1 :2,5 y excipientes farmacéuticamente […]
COMBINACIÓN FARMACÉUTICA SINÉRGICA QUE COMPRENDE TRAMADOL CLORHIDRATO Y PREGABALINA, Y SU USO PARA EL TRATAMIENTO DEL DOLOR NEUROPÁTICO, del 5 de Marzo de 2020, de GRUNENTHAL GMBH: Combinación farmacéutica sinérgica que contiene tramadol clorhidrato y pregabalina en una razón p/p entre 1 :1,5 a 12,5 y excipientes farmaceuticamente aceptables, […]
Composiciones antimicrobianas mejoradas, del 5 de Febrero de 2020, de NOVAPHARM RESEARCH (AUSTRALIA) PTY LTD: Una composición antiséptica para frotado de manos que cuando es usada a una tasa menor que 6 ml de composición durante hasta 60 segundos produce un nivel de eficacia biocida […]