Productos resistentes a manipulaciones indebidas, para la administración de opioides.
Forma farmacéutica de dosificación oral que comprende
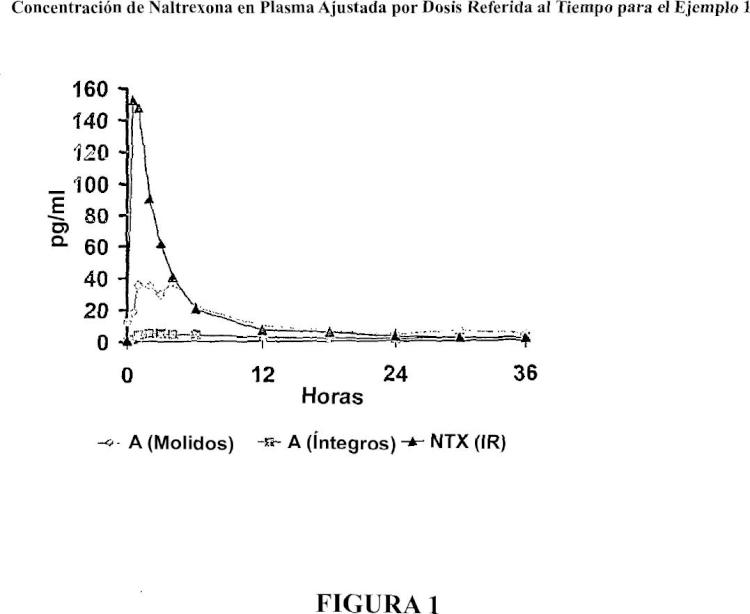
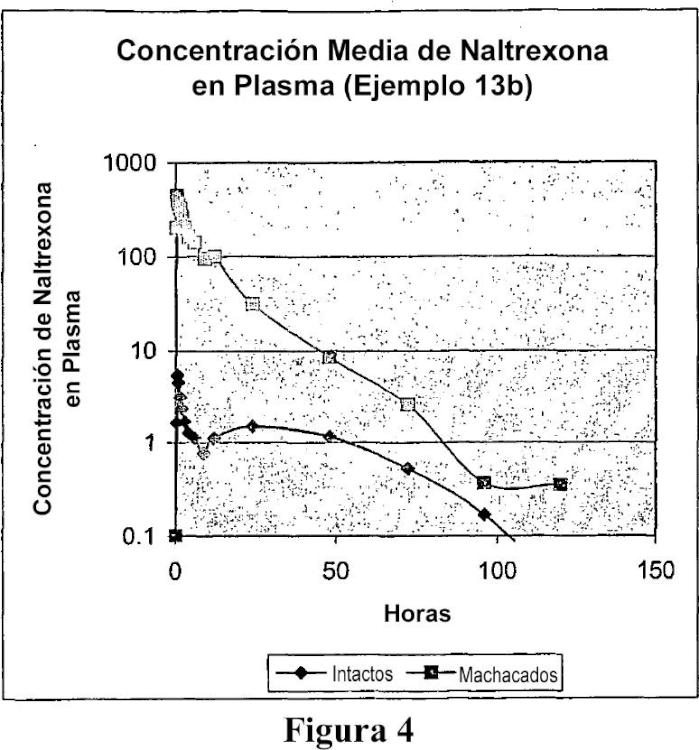
(a) una partícula que comprende un antagonista opioide dispersado en una matriz extruida en fusión,
en donde la matriz comprende entre un 1% y un 80% en peso de uno o más primeros materiales hidrófobos farmacéuticamente aceptables, y
(b) una capa de segundo material hidrófobo que recubre la partícula.
Tipo: Patente Europea. Resumen de patente/invención. Número de Solicitud: E10010924.
Solicitante: EURO-CELTIQUE S.A..
Nacionalidad solicitante: Luxemburgo.
Dirección: 2, AVENUE CHARLES DE GAULLE 1653 LUXEMBOURG LUXEMBURGO.
Inventor/es: OSHLACK, BENJAMIN, CHASIN, MARK, HUANG, HUA-PIN, VAN BUSKIRK,GLENN, VASHI,VIJAY.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A61K31/485 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › Derivados del morfinano, p. ej. morfina, codeína.
- A61K9/14 A61K […] › A61K 9/00 Preparaciones medicinales caracterizadas por un aspecto particular. › en estado especial, p. ej. polvos (microcápsulas A61K 9/50).
- A61P25/04 A61 […] › A61P ACTIVIDAD TERAPEUTICA ESPECIFICA DE COMPUESTOS QUIMICOS O DE PREPARACIONES MEDICINALES. › A61P 25/00 Medicamentos para el tratamiento de trastornos del sistema nervioso. › Analgésicos que actúan sobre el sistema nervioso central, p.ej. opioides.
PDF original: ES-2553203_T3.pdf

Patentes similares o relacionadas:
Regímenes de dosificación de buprenorfina, del 1 de Julio de 2020, de Indivior UK Limited: Buprenorfina para su uso en un método para tratar el trastorno por uso de opioides en un ser humano que lo necesite, que comprende las etapas […]
 Forma de dosificación llena de líquido, disuasoria del abuso y de liberación inmediata, del 24 de Junio de 2020, de Pharmaceutical Manufacturing Research Services, Inc: Una cápsula disuasoria del abuso de liberación inmediata, que comprende:
(a) un principio activo susceptible de abuso;
(b) un primer polietilenglicol (PEG) […]
Forma de dosificación llena de líquido, disuasoria del abuso y de liberación inmediata, del 24 de Junio de 2020, de Pharmaceutical Manufacturing Research Services, Inc: Una cápsula disuasoria del abuso de liberación inmediata, que comprende:
(a) un principio activo susceptible de abuso;
(b) un primer polietilenglicol (PEG) […]
Gránulos de dispersión rápida, comprimidos de desintegración oral y métodos, del 3 de Junio de 2020, de Adare Pharmaceuticals, Inc: Microgránulos de dispersión rápida, farmacéuticamente aceptables, que tienen una mediana del tamaño de partícula en el rango de 100 μm a 300 […]
Pulverizador de buprenorfina sublingual, del 22 de Abril de 2020, de Fresh Cut Development, LLC: Una formulación de pulverización sublingual que comprende una cantidad efectiva de buprenorfina, o una sal farmacéuticamente aceptable de la misma, […]
Dispositivos de administración transdérmica resistentes al abuso y composiciones que comprenden un agonista de opioides y un derivado N-óxido no administrado por vía transdérmica de un antagonista de opioide para el tratamiento del dolor, del 22 de Abril de 2020, de EURO-CELTIQUE S.A.: Un dispositivo de administración transdérmica que comprende una composición farmacéutica, en el que dicha composición comprende un derivado […]
Formulación farmacéutica a base de ibuprofeno y codeína que tiene estabilidad mejorada, del 15 de Abril de 2020, de FARMASIERRA MANUFACTURING S.L: Formulación farmacéutica a base de ibuprofeno y codeínade estabilidad mejorada. La invención consiste en una nueva formulación farmacéuticaen forma […]
Parche transdérmico de oximorfona, del 8 de Abril de 2020, de Buzzz Pharmaceuticals Limited: Un parche transdérmico que tiene una capa que contiene el fármaco que comprende oximorfona o una sal farmacéuticamente aceptable de la misma, […]
Composiciones de buprenorfina y antagonistas del receptor de opioides mu, del 1 de Abril de 2020, de Alkermes Pharma Ireland Limited: Una composición que comprende buprenorfina y un antagonista del receptor opioide μ, en donde la composición está caracterizada por un Índice […]