Procedimientos e intermedios para la preparación de un inhibidor macrocíclico de la proteasa de VHC.
Un procedimiento para preparar el compuesto de fórmula (VIII),
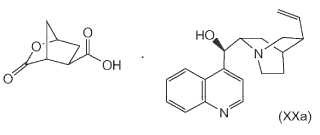
partiendo de una sal de cinconidina (XXa), que sehace reaccionar con N-metil-hexenamina (NMHA) (XIX) en una reacción de formación de amida para producir laamida de la lactona bicíclica (XVIII), en la que el grupo lactona se abre para producir el producto deseado (VIII),como se ilustra en el esquema a continuación, en el que R1 es alquilo de C1-4:**Fórmula**
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/EP2009/067715.
Solicitante: Janssen Pharmaceuticals, Inc.
Nacionalidad solicitante: Estados Unidos de América.
Dirección: 1125 TRENTON-HARBOURTON ROAD TITUSVILLE, NJ 08560 ESTADOS UNIDOS DE AMERICA.
Inventor/es: HORVATH, ANDRAS, DEPRE,DOMINIQUE PAUL MICHEL, ORMEROD,DOMINIC JOHN, CERPENTIER,VÉRONIQUE.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C07D311/06 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07D COMPUESTOS HETEROCICLICOS (Compuestos macromoleculares C08). › C07D 311/00 Compuestos heterocíclicos que contienen ciclos de seis miembros que contienen un átomo de oxígeno como único heteroátomo, condensados con otros ciclos. › con átomos de oxígeno o azufre directamente unidos en posición 2.
- C07D417/04 C07D […] › C07D 417/00 Compuestos heterocíclicos que contienen dos o más heterociclos, teniendo al menos un ciclo átomos de nitrógeno y azufre como únicos heteroátomos del ciclo, no previstos por el C07D 415/00. › unidos directamente por un enlace entre dos miembros cíclicos.
- C07D453/04 C07D […] › C07D 453/00 Compuestos heterocíclicos que contienen sistemas cíclicos de quinuclidina o isoquinuclidina, p. ej. alcaloides de quinina. › teniendo unido en posición 2 un radical quinolil-4, un radical quinolil-4 sustituido o un radical alquilendioxi quinolil-4 unido por un solo átomo de carbono, p. ej. quinina.
PDF original: ES-2429013_T3.pdf
Fragmento de la descripción:
Procedimientos e intermedios para la preparación de un inhibidor macrocíclico de la proteasa de VHC
Campo de la invención La presente invención se refiere a procedimientos de síntesis e intermedios de síntesis de un inhibidor macrocíclico de la proteasa del virus de la hepatitis C (VHC) .
Antecedentes de la invención El virus de la hepatitis C (VHC) es la causa principal de hepatitis crónica, que puede progresar a fibrosis hepática conducente a cirrosis, enfermedad hepática de fase final, y HCC (carcinoma hepatocelular) , haciendo del mismo la causa principal de los trasplantes de hígado. La terapia actual anti-VHC, basada en interferón-alfa (IFN-!) (pegilado) en combinación con ribavirina, sufre de una eficacia limitada, efectos secundarios importantes, y es deficientemente tolerada en muchos pacientes. Esto ha alentado la búsqueda de una terapia más eficaz, conveniente y mejor tolerada.
La replicación del genoma de VHC está mediada por varias enzimas, entre las cuales se encuentra la serinaproteasa NS3 del VHC y su cofactor asociado, NS4A. Se han descrito diversos agentes que inhiben esta enzima. El documento WO 05/073195 describe inhibidores lineales y macrocíclicos de la serina-proteasa NS3 con un resto de prolina central sustituido, y el documento WO 05/073216 con un resto de ciclopentilo central. Entre éstos, los derivados macrocíclicos son atractivos en el sentido de que exhiben una notable actividad contra VHC y un perfil farmacocinético satisfactorio.
El documento WO 2007/014926 describe derivados ciclopentílicos y de prolina macrocíclicos que incluyen el compuesto de fórmula (I) , con la estructura representada en lo sucesivo. El compuesto de fórmula (I) es un inhibidor muy eficaz de la serina proteasa del VHC, y es particularmente atractivo debido a su perfil farmacocinético favorable. Debido a sus propiedades, este compuesto se ha seleccionado como candidato potencial para el desarrollo de un fármaco contra el VHC. En consecuencia, existe la necesidad de producir grandes cantidades de este ingrediente activo basándose en procedimientos que proporcionan el producto con rendimiento elevado y con un grado elevado de pureza. El documento WO 2008/092955 describe procedimientos e intermedios para preparar el compuesto de fórmula (I) .
El compuesto de fórmula (I) se puede preparar partiendo de un intermedio (VI) , en el que la función éster se hidroliza, produciendo ácido carboxílico (V) , que a su vez se acopla en una reacción de formación de amida con el ciclopropilaminoácido (Va) . El intermedio resultante (IV) se cicla mediante una reacción de metátesis de olefinas en presencia de un catalizador metálico adecuado, tal como, por ejemplo, un catalizador ilidénico a base de Ru. El éster macrocíclico resultante (III) se hidroliza entonces al ácido macrocíclico (II) . Este último se acopla con una sulfonilamida (V) en una reacción de formación de amida para producir el producto final (I) . Estas reacciones se esquematizan en el esquema de reacción aquí más abajo. En este y en los siguientes esquemas de reacción o representaciones de compuestos individuales, R es alquilo de C1-4, en particular R es alquilo de C1-3, más en particular, R es alquilo de C1-2, o en una realización, R es etilo. R1 es alquilo de C1-4, en particular R1 es alquilo de C13, más en particular, R1 es alquilo de C1-2, o R1 es metilo; o R1 es etilo.
A su vez, el intermedio (VI) se puede preparar usando procedimientos descritos en el documento WO 2008/092955, en particular partiendo de un bis-éster hidroxiciclopentílico de fórmula (Xa) ,
(a) haciendo reaccionar el bis-éster hidroxiciclopentílico de fórmula (Xa) con un quinolinol sustituido con tiazolilo (VII) en una reacción de formación de éter, obteniendo así un bis-éster quinoliniloxiciclopentílico de fórmula (XII) , en el que el grupo éster bencílico que está en posición cis con respecto al grupo éter en el biséster quinoliniloxi-ciclopentílico de fórmula (XII) se escinde selectivamente a un ácido monocarboxílico (XI) , que a su vez se acopla con una alquenilamina en una reacción de formación de amida, obteniendo así el producto final deseado de fórmula (VI) ; o (b) convertir selectivamente el bis-éster hidroxiciclopentílico de fórmula (Xa) al ácido monocarboxílico (IX) , que a su vez se acopla con una alquenilamina en una reacción de formación de amida para obtener hidroxiciclopentilamida (VIII) , que a su vez se hace reaccionar con un quinolinol sustituido con tiazolilo (VII) , obteniendo así el producto final deseado de fórmula (VI) ; como se esquematiza en el siguiente esquema de reacción:
Cada R1 en los procedimientos representados en el esquema anterior es como se especifica anteriormente, y preferiblemente R1 es metilo. Bn representa bencilo.
La presencia de diversos centros quirales en el compuesto de fórmula (I) y sus predecesores plantea retos particulares por cuanto la pureza quiral es esencial para tener un producto que sea aceptable para uso terapéutico. El intermedio (VI) tiene tres centros quirales, y la obtención de la estereoquímica correcta para los tres centros es un reto importante para cualesquiera procedimientos de síntesis dirigidos a preparar este compuesto. Por tanto, los procedimientos para preparar (VI) deberían dar como resultado productos de pureza quiral aceptable sin el uso de procedimientos de purificación engorrosos con la pérdida de cantidades sustanciales de formas estereoisómeras indeseadas.
El documento WO 2008/092955 describe un procedimiento de síntesis para el intermedio (Xa) partiendo de ácido 4ºxo-ciclopentil-1, 2-bis-carboxílico (XVII) reduciendo la función ceto a un alcohol, obteniendo así ácido 4-hidroxi
ciclopentil-1, 2-bis-carboxílico (XVI) , que a su vez se cicla a la lactona bicíclica (XV) , en la que el grupo ácido carboxílico en la lactona bicíclica (XV) se esterifica con alcohol bencílico obteniendo así el éster bencílico de la lactona (XIV) . La lactona en este último se abre mediante una reacción de transesterificación en presencia de un alcanol de C1-4, produciendo así el bis-éster hidroxiciclopentílico de fórmula (X) , que a su vez se resuelve en los enantiómeros (Xa) y (Xb) ; como se esquematiza en el siguiente esquema de reacción:
Cada R1 en los procedimientos representados en el esquema anterior es como se especifica anteriormente, y preferiblemente R1 es metilo.
Una desventaja del procedimiento anterior es que implica una resolución de los enantiómeros de (X) mediante cromatografía en columna quiral, un procedimiento engorroso que es difícil de realizar en una producción a gran escala.
Honda et al., Tetrahedron Letters, vol. 22, no. 28, p. 2679-2682, 1981, describen la síntesis de la (±) -brefeldina A usando los siguientes materiales de partida:
La síntesis de Honda et al. parte de ácido dl-trans-4-oxociclopentano-1, 2-dicarboxílico 2, que se esterificó para dar el éster metílico correspondiente 3, y se redujo con Ni Raney al alcohol 4. La hidrólisis parcial de 4 al ácido monocarboxílico y la bencilación con bromuro de bencilo dieron predominantemente el diastereoisómero 5, a saber, el diastereoisómero en el cual los grupos hidroxi y éster bencílico se encuentran en posición cis. Este último éster 5 en Honda et al. y el compuesto (X) son ambos racematos, pero son diastereoisómeros entre sí, más precisamente epímeros en el carbono nº 4 que lleva el grupo hidroxi. El compuesto (Xa) es uno de los dos enantiómeros obtenidos por separación del compuesto racémico (X) . El otro enantiómero es el compuesto (Xb) .
El documento WO 2005/073195 describe la síntesis de la lactona bicíclica (8b) enantioméricamente pura a partir de un enantiómero de 3, 4-bis (metoxicarbonil) ciclopentanona. Esta última se preparó como se describió por Rosenquist et al. en Acta Chemica Scandinavica 46 (1992) 1127-1129. El isómero trans- (3R, 4R) -3, 4bis (metoxicarbonil) ciclopentanona se convirtió en la lactona bicíclica (8b) :
El documento WO 2005/073195 describe adicionalmente una modificación ulterior de la lactona (8b) al éster de t.Bu por apertura de la lactona y acoplamiento con aminoácidos adecuadamente protegidos, v.g. con el éster etílico del ácido (1R, 2S) -1-amino-2-vinilciclopropano-carboxílico, que el último caso proporciona:
La formación de los compuestos de fórmula (I) implica necesariamente introducir en el anillo de ciclopentilo el resto de quinolina sustituido con tiazolilo, vía un enlace de éter. La reacción de Mitsunobu ofrece una ruta de reacción 15 atractiva para la preparación de éteres de alquilo aromáticos en los cuales un éter de alquilo se activa y se hace reaccionar con un fenol. Adicionalmente, las... [Seguir leyendo]
Reivindicaciones:
1. Un procedimiento para preparar el compuesto de fórmula (VIII) , partiendo de una sal de cinconidina (XXa) , que se hace reaccionar con N-metil-hexenamina (NMHA) (XIX) en una reacción de formación de amida para producir la amida de la lactona bicíclica (XVIII) , en la que el grupo lactona se abre para producir el producto deseado (VIII) , como se ilustra en el esquema a continuación, en el que R1 es alquilo de C1-4:
2. El procedimiento de la reivindicación 1, en el que R1 es metilo.
3. El procedimiento de las reivindicaciones 1 ó 2, en el que la reacción de formación de amida se lleva a cabo en 10 presencia de un reactivo de acoplamiento de amida en un disolvente inerte para la reacción.
4. Un procedimiento como en la reivindicación 3, en el que la reacción se lleva a cabo en presencia de una base.
5. El procedimiento de la reivindicación 3 o reivindicación 4, en el que el disolvente comprende hidrocarburos halogenados, éteres, alcoholes, disolventes hidrocarbonados, disolventes apróticos dipolares, o sus mezclas.
6. El procedimiento de la reivindicación 5, en el que los disolventes halogenados son diclorometano (DCM) o
cloroformo, los éteres son tetrahidrofurano (THF) o 2-metiltetrahidrofurano (MeTHF) , los alcoholes son metanol o etanol, los disolventes hidrocarbonados son tolueno o xileno, los disolventes apróticos dipolares son DMF, DMA, acetonitrilo.
7. El procedimiento de la reivindicación 3 o reivindicación 4, en el que los agentes de formación de amida comprenden agentes tales como N-etoxicarbonil-2-etoxi-1, 2-dihidroquinolina (EEDQ) , N-isopropoxicarbonil-2
isopropoxi-1, 2-dihidroquinolina (IIDQ) , hexafluorofosfato de N, N, N’, N’-tetrametil-O- (7-azabenzotriazol-1-il) uronio (HATU) , hexafluorofosfato de benzotriazol-1-il-oxi-tris-pirrolidino-fosfonio, CDI, 1-etil-3- (3dimetilaminopropil) carbodiimida (EDCI) o su hidrocloruro, diciclohexilcarbodiimida (DCC) , 1, 3diisopropilcarbodiimida, o hexafluorofosfato de O-benzotriazol-N, N, N’, N’-tetrametil-uronio (HBTU) .
8. El procedimiento de la reivindicación 7, en el que el agente de formación de amida está en presencia de un 25 catalizador.
9. El procedimiento de la reivindicación 8, en el que el catalizador es 1-hidroxibenzotriazol (HOBt) o 4dimetilaminopiridina (DMAP) .
10. El procedimiento de la reivindicación 4, en el que la base es una amina terciaria.
11. El procedimiento de la reivindicación 10, en el que la amina terciaria es trietilamina, N-metilmorfolina, N, N30 diisopropiletilamina.
12. Un procedimiento para preparar la sal de cinconidina (XXa) , que se obtiene de la sal racémica (XX) mediante cristalización:
13. El procedimiento de la reivindicación 12, en el que la sal racémica (XX) se obtiene poniendo en contacto el ácido carboxílico de la lactona bicíclica (XV) con cinconidina:
14. El procedimiento de la reivindicación 13, en el que se añade una suspensión de cinconidina a una disolución de (XV) a una temperatura de alrededor de 50 a alrededor de 70ºC, y posteriormente se permite que la mezcla se enfríe, con lo que el producto deseado (XXa) cristaliza.
15. El procedimiento de la reivindicación 13, en el que (XV) se disuelve en un disolvente seleccionado de disolventes de tipo éster, y los disolventes para la suspensión de cinconidina incluyen acetonitrilo.
16. El procedimiento de la reivindicación 15, en el que los disolventes de tipo éster son acetato de etilo.
17. El procedimiento de las reivindicaciones 14 or 15, en el que la formación de sal se lleva a cabo a una temperatura de alrededor de 60ºC, y la mezcla se deja enfriar hasta alrededor de la temperatura ambiente.
18. El procedimiento de la reivindicación 17, en el que la mezcla se deja enfriar hasta una temperatura en el intervalo de alrededor de 20 a alrededor de 25ºC.
19. El procedimiento de las reivindicaciones 14 ó 15, en el que la sal se purifica adicionalmente mediante recristalización en un disolvente apropiado o mezcla de disolventes, o resuspendiendo en un disolvente o mezcla de disolventes.
20. El procedimiento de la reivindicación 19, en el que el disolvente en la recristalización es un alcanol de C1-4, o en la resuspensión el disolvent o mezcla de disolventes es una mezcla de etanol/agua.
21. El procedimiento de la reivindicación 20, en el que el alcanol de C1-4 es isopropanol, y/o la mezcla de etanol/agua es una mezcla de etanol/agua 5%/95% (p/p) .
22. La sal de cinconidina de fórmula
23. El uso de la sal de cinconidina (XXa) definida en la reivindicación 22, como intermedio en la preparación del intermedio (VIII) .
24. Un procedimiento para la preparación de un compuesto de fórmula (I)
procedimiento el cual comprende un procedimiento para la preparación de un compuesto de fórmula (VIII) según una cualquiera de las reivindicaciones 1 a 11, seguido de la conversión en un compuesto de fórmula (I) .
25. Un procedimiento como en la reivindicación 24, en el que la conversión al compuesto de fórmula (I) es como sigue:
(i) hacer reaccionar un compuesto de fórmula (VIII) con un compuesto de fórmula (VII) para formar un compuesto de fórmula (VI) ,
(ii) convertir (VI) en (I) según el siguiente esquema:
26. Un procedimiento para la preparación de un compuesto de fórmula (I) , como se define en la reivindicación 24, procedimiento el cual comprende un procedimiento para la preparación de un compuesto de fórmula (XXa) según una cualquiera de las reivindicaciones 12 a 21, seguido de la conversión en un compuesto de fórmula (I) .
27. Un procedimiento como en la reivindicación 26, en el que el compuesto de fórmula (XXa) se convierte primero en un compuesto de fórmula (VIII) siguiendo un procedimiento según una cualquiera de las reivindicaciones 1 a 11.
28. Un procedimiento como en la reivindicación 27, en el que la conversión del compuesto de fórmula (VIII) al compuesto de fórmula (I) es según la reivindicación 25.
29. El uso de la sal de cinconidina (XXa) definida en la reivindicación 22, como intermedio en la preparación del 10 compuesto (I) como se define en la reivindicación 24.
Patentes similares o relacionadas:
Compuestos de heteroaril carboxamida como inhibidores de RIPK2, del 29 de Julio de 2020, de BOEHRINGER INGELHEIM INTERNATIONAL GMBH: Un compuesto de fórmula (I): **(Ver fórmula)** o sus sales farmacéuticamente aceptables, en la que: X es N y Y es CH; o X es CH y Y es N; […]
Composiciones y métodos anti-HCMV, del 22 de Julio de 2020, de Evrys Bio, LLC: Una composición que comprende un compuesto de la Fórmula I: **(Ver fórmula)** en donde: Ar es **(Ver fórmula)** en donde cada anillo cíclico de 5 o 6 miembros […]
Compuestos de indol carboxamida útiles como inhibidores de cinasas, del 13 de Mayo de 2020, de BRISTOL-MYERS SQUIBB COMPANY: Un compuesto de la fórmula (I): **(Ver fórmula)** o una sal del mismo, en la que: X es CR4; A es: **(Ver fórmula)** o **(Ver fórmula)** Q2 es […]
Compuestos monocíclicos sustituidos con heteroarilo, del 29 de Abril de 2020, de BRISTOL-MYERS SQUIBB COMPANY: Un compuesto de Fórmula (I): **(Ver fórmula)** o una de sus sales; en la que: R1 es -OCH3, -OCHF2 o -CH2OCH3; R2 es H, F, Cl, Br, -10 OH, […]
Derivados de 1-ciano-pirrolidina como inhibidores de USP30, del 29 de Abril de 2020, de Mission Therapeutics Limited: Un compuesto que tiene la fórmula (I): **(Ver fórmula)** un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde: m […]
Piridinas sustituidas con heteroarilo y métodos de uso, del 22 de Abril de 2020, de AbbVie Overseas S.à r.l: Un compuesto de Formula I, o una sal farmaceuticamente aceptable del mismo, **(Ver fórmula)** en donde X1 y X2 se seleccionan independientemente de H; […]
Compuestos de atropisómeros tricíclicos, del 15 de Abril de 2020, de BRISTOL-MYERS SQUIBB COMPANY: Un compuesto de fórmula (I) **(Ver fórmula)** o una de sus sales, en la que: las dos líneas de puntos representan dos enlaces sencillos o dos […]
Compuestos de trifluoroalquenilo heterocíclicos que tienen actividad nematicida, sus composiciones agronómicas y uso de los mismos, del 8 de Abril de 2020, de ISAGRO S.P.A.: Compuestos de trifluoroalquenilo heterocíclicos que tienen Fórmula (I): Het-S(O)n-(CH2)m-CF=CF2 (I) en la que: - Het representa un grupo heterocíclico aromático, […]