PROCEDIMIENTO PARA LA PRODUCCIÓN DE LA L-LISINA QUE UTILIZA ESCHERICHIA COLI.
Procedimiento para producir L-lisina que utiliza una Escherichia coli que comprende las etapas que consisten en cultivar la Escherichia coli en un medio para producir y acumular la L-lisina en el medio y recoger la L-lisina,
en el que la Escherichia coli es una cepa mutante o una cepa recombinante genética en la que la actividad de la citocromo oxidasa de tipo bo es aumentada en comparación con la cepa sin modificar mediante el incremento del número de copias de un gen que codifica la citocromo oxidasa de tipo bo, la modificación de una secuencia reguladora de la expresión del gen, o su combinación
Tipo: Patente Europea. Resumen de patente/invención. Número de Solicitud: E09003058.
Solicitante: AJINOMOTO CO., INC..
Nacionalidad solicitante: Japón.
Dirección: 15-1, KYOBASHI 1-CHOME CHUO-KU TOKYO 104-8315 JAPON.
Inventor/es: ITO, HISAO, KURAHASHI, OSAMU, NAKAI,YUTA, NAKANISHI,KAZUO, KAWAHARA,YOSHIO.
Fecha de Publicación: .
Fecha Solicitud PCT: 2 de Julio de 2001.
Clasificación Internacional de Patentes:
- C12N9/02D
- C12N9/02D2
- C12P13/08 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12P PROCESOS DE FERMENTACION O PROCESOS QUE UTILIZAN ENZIMAS PARA LA SINTESIS DE UN COMPUESTO QUIMICO DADO O DE UNA COMPOSICION DADA, O PARA LA SEPARACION DE ISOMEROS OPTICOS A PARTIR DE UNA MEZCLA RACEMICA. › C12P 13/00 Preparación de compuestos orgánicos que contienen nitrógeno. › Lisina; Acido diaminopimélico; Treonina; Valina.
- C12P13/22 C12P 13/00 […] › Triptófano; Tirosina; Fenilalanina; 3,4-Dihidroxifenilalanina.
Clasificación PCT:
- C12P13/08 C12P 13/00 […] › Lisina; Acido diaminopimélico; Treonina; Valina.
Países PCT: Austria, Bélgica, Suiza, Alemania, Dinamarca, España, Francia, Reino Unido, Grecia, Italia, Liechtensein, Luxemburgo, Países Bajos, Suecia, Mónaco, Portugal, Irlanda, Finlandia, Chipre.
PDF original: ES-2360174_T3.pdf
Fragmento de la descripción:
Antecedentes de la invención
Campo de la invención
La presente invención se refiere a un procedimiento para producir una sustancia que utiliza un microorganismo. En la presente invención, el microorganismo es la Escherichia coli. La sustancia que va a producirse es la L-lisina.
Descripción de la técnica relacionada
Muchos organismos adquieren energía para las actividades vitales mediante la respiración. En la respiración de los microorganismos, diversos complejos enzimáticos generalmente funcionan según la especie o ambiente de crecimiento, y la eficiencia de captación de la energía también varía significativamente. Los carbohidratos, proteínas y ácidos alifáticos se convierten en acetil-CoA en la glicólisis, en la β-oxidación y similares, y se descomponen en el ciclo del ácido cítrico. La energía conservada en forma de NADH se utiliza para la excreción de protones de las células microbianas con la ayuda de la NADH deshidrogenasa (NDH) y el posterior sistema de transferencia de electrones constituido por oxidorreductasas, y de esta manera se forma un gradiente de concentración de protones entre el interior y el exterior de la membrana citoplasmática. Este gradiente de concentración de protones se utiliza como fuerza motriz para la síntesis de adenosina trifosfato (ATP). En la actualidad existe una ruta que muestra una capacidad de elevada excreción de protones y una ruta que muestra una capacidad de excreción baja de protones entre las rutas de transferencia de electrones, dependiendo de la combinación de NDH y oxidorreductasas. Se considera que una ruta con capacidad elevada de excreción de proteínas muestra una eficiencia energética elevada y que una ruta de capacidad baja de excreción de protones muestra una eficiencia energética baja. De esta manera, un tipo de microorganismo contiene simultáneamente una pluralidad de rutas respiratorias de cadena de transferencia de electrones en paralelo, y entre estas rutas se incluyen las de eficiencia energética elevada y las de baja eficiencia energética.
Existen dos tipos de NDH y dos tipos de oxidasas terminales en la cadena respiratoria de Escherichia coli para un estado aeróbico. Es decir, respecto a NDH, son conocidos NDH-1 (codificada por el operón nuo) de eficiencia energética elevada, y NDH-II (codificado por ndh) de eficiencia energética baja. Además, respecto a la oxidasa terminal, es conocida la citocromo oxidasa tipo bo (codificada por el operón cyoABCD), clasificada en tipo SoxM (Castresana, J. y Saraste, M., Trends in Biochem. Sci. 20:443-448, 1995) y que muestra eficiencia energética elevada, y la citocromo oxidasa tipo bd (codificada por cydAB), que muestra una eficiencia energética baja. Aunque es conocido que las cantidades de expresión de estos enzimas de cadena respiratoria varían en respuesta al ambiente de crecimiento (Minagawa et al., The Journal of Biological Chemistry 265:11198-11203, 1990; Tseng et al., Journal of Bacteriology 178:1094-1098, 1996; Green et al., Molecular Microbiology 12:433-444, 1994; Bongaerts et al., Molecular Microbiology 16:521-534, 1995), todavía existen muchos aspectos desconocidos sobre el significado fisiológico de los patrones de expresión de los mismos.
Además, en Corynebacterium glutamicum existe un complejo citocromo bc1 y se ha confirmado la presencia de por lo menos dos tipos de oxidasa terminal: la oxidasa de tipo SoxM y la citocromo oxidasa de tipo bd (The Second Symposium Concerning Metabolic Engineering, Lecture Abstracts, 1999). Lo expuesto anteriormente demuestra que la ruta de transferencia de electrones desde el reservorio de quinona hasta la molécula de oxígeno incluye dos tipos de ruta: una ruta que utiliza el complejo citocromo bc1 y la oxidasa de tipo SoxM, y una ruta que utiliza únicamente la citocromo oxidasa de tipo bd. Se considera que la primera es una ruta de transferencia de electrones de eficiencia energética elevada, en la que el valor de transferencia de protones para la transferencia de un electrón es reducido.
Respecto a la oxidasa terminal de E. coli, si se compara el rendimiento de crecimiento en un cultivo aeróbico de una cepa mutante que presenta únicamente la citocromo oxidasa de tipo bo, de una cepa mutante que presenta únicamente la citocromo oxidasa de tipo bd y de una cepa salvaje que presenta ambas, el rendimiento de crecimiento es más bajo en la cepa mutante que presenta únicamente la citocromo oxidasa de tipo bd, y depende del tipo y eficiencia de captación energética de la oxidasa terminal (Annual Meeting of the Society for fermentation and Bioengineering Japón, 1995, Lecture Abstracts, nº 357).
Además, se ha informado de la eficiencia energética de mutantes deficientes en algunos enzimas de la cadena respiratoria (Calhoun et al., Journal of Bacteriology 175:3020-3925, 1993).
Sin embargo, no se ha informado de cambios de la eficiencia energética mediante amplificación de un gen de la cadena respiratoria que proporciona eficiencia elevada, tal como aquellos para la oxidasa de tipo NDH-I y de tipo SoxM, e incluso no se conocen intentos para la utilización del mismo en la producción de sustancias. Además, no se han realizado intentos para utilizar la deleción de un enzima de cadena respiratoria de eficiencia baja, tal como NDHII y la citocromo oxidasa de tipo bd, en la producción de sustancias.
Sumario de la invención
Se requiere energía para la biosíntesis de sustancias, tales como L-aminoácidos y ácidos nucleicos en seres vivos. La mayor parte de la energía utilizada consta de poder reductor de NDH, NADPH y similares, y de la energía conservada en forma de ATP. Por lo tanto, en el contexto de la presente invención se ha concebido que, si se incrementa el suministro de energía utilizado en la producción de sustancias diana en los procedimientos para producir sustancias diana mediante la utilización de microorganismos, se mejoraría la productividad de las sustancias diana. Basándose en este concepto, un objetivo de la presente invención es concebir un microorganismo que muestre una eficiencia energética mejorada y proporcionar un procedimiento para producir una sustancia diana mediante la utilización del mismo.
En el contexto de la presente invención se ha concebido que podría construirse un microorganismo que muestre un suministro energético incrementado, aumentando una ruta de cadena respiratoria que muestre una eficiencia elevada de captación energética o haciendo deficiente la ruta de cadena respiratoria que muestra una eficiencia baja de captación energética. Específicamente, respecto a E. coli, se prepararon cepas que se considera que presentan una eficiencia energética mejorada, mediante la amplificación de un gen codificante de una citocromo oxidasa de tipo bo a modo de enzima de cadena respiratoria de eficiencia energética elevada, o delecionando un gen codificante de NDH-II a modo de enzima de cadena respiratoria de eficiencia energética baja. A continuación, se llevó a cabo la producción de L-aminoácidos mediante la utilización de dichas cepas y se encontró que la productividad de los L-aminoácidos se encontraba mejorada en las cepas en las que se había mejorado la eficiencia energética. De esta manera se consiguió la presente invención.
Es decir, la presente invención proporciona lo siguiente:
(1) Un procedimiento para producir la L-lisina utilizando una Escherichia coli que comprende las etapas que consisten en cultivar la Escherichia coli en un medio para producir y acumular la L-lisina en el medio y recoger la Llisina, en el que la Escherichia coli es una cepa mutante o una cepa recombinante genética en la que la actividad de la citocromo oxidasa de tipo bo es aumentada en comparación con la cepa sin modificar mediante el incremento del número de copias de un gen que codifica la citocromo oxidasa de tipo bo, la modificación de una secuencia reguladora de la expresión del gen, o su combinación.
(2) El procedimiento según (1), en el que dicha secuencia reguladora de la expresión es un promotor.
(3) El procedimiento según (1) o (2), en el que dicha citocromo oxidasa de tipo bo se encuentra codificada por el operón cyo.
Según la presente invención, puede mejorarse la productividad de la sustancia diana basándose en un principio diferente del utilizado en la estrategia convencional.
Breve explicación de los dibujos
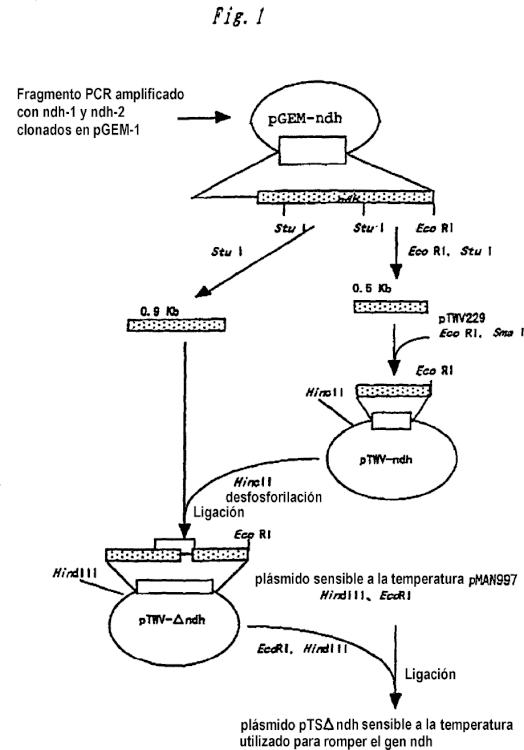
La figura 1 muestra la construcción del plásmido pTS-Δndh para producir una cepa con alteración del gen NDH-II.
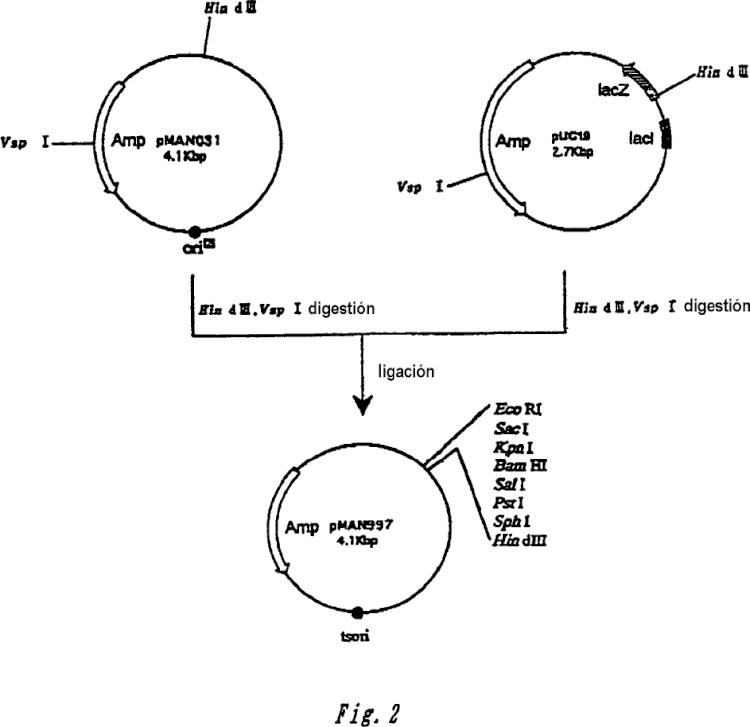
La figura 2 muestra la construcción de pMAN997.
Descripción detallada de la... [Seguir leyendo]
Reivindicaciones:
1. Procedimiento para producir L-lisina que utiliza una Escherichia coli que comprende las etapas que consisten en cultivar la Escherichia coli en un medio para producir y acumular la L-lisina en el medio y recoger la L-lisina, en el que la Escherichia coli es una cepa mutante o una cepa recombinante genética en la que la actividad de la citocromo oxidasa de tipo bo es aumentada en comparación con la cepa sin modificar mediante el incremento del número de copias de un gen que codifica la citocromo oxidasa de tipo bo, la modificación de una secuencia reguladora de la expresión del gen, o su combinación.
10 2. Procedimiento según la reivindicación 1, en el que dicha secuencia reguladora de la expresión es un promotor.
3. Procedimiento según la reivindicación 1 ó 2, en el que dicha citocromo oxidasa de tipo bo se encuentra codificada por el operón cyo.
Patentes similares o relacionadas:
Corynebacterium, del 15 de Julio de 2020, de CJ CHEILJEDANG CORPORATION: Un microorganismo productor de L-lisina del género Corynebacterium en donde una proteína que comprende una secuencia de aminoácidos de la […]
Microorganismo del género Corynebacterium de producción de lisina y procedimiento de producción de lisina usando el mismo, del 10 de Junio de 2020, de CJ CHEILJEDANG CORPORATION: Un microorganismo del género Corynebacterium productor de L-lisina, en el que la vía de biosíntesis de L-lisina está potenciada y la oxalacetato-descarboxilasa […]
Método para la producción de L-aminoácido, del 3 de Junio de 2020, de AJINOMOTO CO., INC.: Método para producir un L-aminoácido mediante fermentación que comprende cultivar un microorganismo que tiene la capacidad de producción de L-aminoácido en […]
Microorganismo con productividad de l-lisina aumentada y procedimiento para producir l-lisina utilizando el mismo, del 27 de Mayo de 2020, de CJ CHEILJEDANG CORPORATION: Una subunidad beta prima (subunidad-β') mutante de la ARN polimerasa, en la que la subunidad beta prima (subunidad-β') mutante de la ARN polimerasa tiene […]
Microorganismo que tiene una productividad aumentada de l-lisina y método para producir l-lisina mediante el uso del mismo, del 8 de Abril de 2020, de CJ CHEILJEDANG CORPORATION: Un microorganismo productor de L-lisina del género Corynebacterium en el que al menos una proteína secretora seleccionada del grupo que consiste […]
Método para producir una sustancia diana mediante un procedimiento de fermentación, del 1 de Abril de 2020, de KYOWA HAKKO BIO CO., LTD: Procedimiento para producir una sustancia diana, que comprende: cultivar, en un medio, una bacteria corineforme en la que la actividad de la proteína FruK y la proteína FruA […]
Microorganismo que produce L-lisina y método para producir L-lisina usando el mismo, del 1 de Abril de 2020, de CJ CHEILJEDANG CORPORATION: Un microorganismo del género Corynebacterium que tiene una mayor productividad de L-lisina en comparación con un microorganismo sin modificar, el cual se modifica de […]
Microorganismo del género corynebacterium para producir l-aminoácido y método para producir l-aminoácido usando el mismo, del 18 de Marzo de 2020, de CJ CHEILJEDANG CORPORATION: Un microorganismo productor de L-lisina del género Corynebacterium en donde una proteína que comprende una secuencia de aminoácidos de la SEQ ID NO: 1 está inactivada.