Peptidomiméticos en horquilla b fijados sobre una plantilla, con actividad antagonista con respecto a CXCR4.
Compuesto de fórmula**Fórmula**
en la que**Fórmula**
es un residuo de dipéptido constituido por dos elementos constructivos de aminoácidos diferentes,
siendo el dipéptido DPro-LPro; y Z es una cadena constituida por 12 residuos de a-aminoácidos, en la que
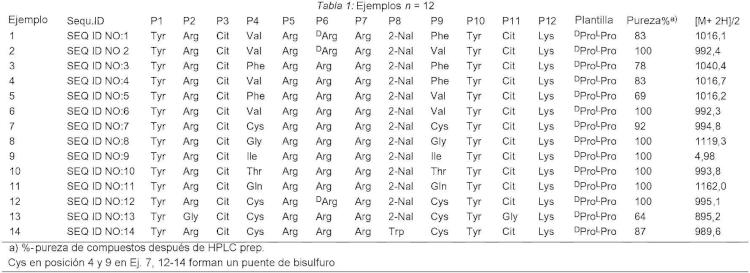
- el residuo P1 es el residuo de Tyr;
- el residuo P2 es el residuo de Arg o Gly;
- el residuo P3 es el residuo de Cit;
- el residuo P4 es el residuo de Val, Phe, Cys, Gly Ile, Thr o Gln;
- el residuo P5 es el residuo de Arg;
- el residuo P6 es el residuo de DArg o Arg;
- el residuo P7 es el residuo de Arg;
- el residuo P8 es el residuo de 2-Nal o Trp;
- el residuo P9 es el residuo de Phe, Val, Cys, Gly, Ile, Thr o Gln;
- el residuo P10 es el residuo de Tyr;
- el residuo P11 es el residuo de Cit o Gly;
- el residuo P12 es el residuo de Lys;
Cys en P4 y P9 forman un puente de bisulfuro
en forma libre o en forma de una sal farmacéuticamente aceptable.
Tipo: Patente Europea. Resumen de patente/invención. Número de Solicitud: E12185905.
Solicitante: Polyphor AG.
Nacionalidad solicitante: Suiza.
Dirección: HEGENHEIMERMATTWEG 125 4123 ALLSCHWIL SUIZA.
Inventor/es: VRIJBLOED, JAN, WIM, OBRECHT, DANIEL, LUDIN, CHRISTIAN, LOCIURO, SERGIO, GOMBERT, FRANK, ZUMBRUNN,JURG, MUKHERJEE,RESHMI, ROBINSON,JOHN,ANTHONY, ROMAGNOLI,BARBARA, HENZE,HEIKO, DEMARCO,Steve J, MÖHLE,KERSTIN.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A61K38/12 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 38/00 Preparaciones medicinales que contienen péptidos (péptidos que contienen ciclos beta-lactama A61K 31/00; dipéptidos cíclicos que no tienen en su molécula ningún otro enlace peptídico más que los que forman su ciclo, p. ej. piperazina 2,5-dionas, A61K 31/00; péptidos basados en la ergolina A61K 31/48; que contienen compuestos macromoleculares que tienen unidades aminoácido repartidas estadísticamente A61K 31/74; preparaciones medicinales que contienen antígenos o anticuerpos A61K 39/00; preparaciones medicinales caracterizadas por los ingredientes no activos, p. ej. péptidos como soportes de fármacos, A61K 47/00). › Péptidos cíclicos.
- C07K1/04 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07K PEPTIDOS (péptidos que contienen β -anillos lactamas C07D; ipéptidos cíclicos que no tienen en su molécula ningún otro enlace peptídico más que los que forman su ciclo, p. ej. piperazina diones-2,5, C07D; alcaloides del cornezuelo del centeno de tipo péptido cíclico C07D 519/02; proteínas monocelulares, enzimas C12N; procedimientos de obtención de péptidos por ingeniería genética C12N 15/00). › C07K 1/00 Procedimientos generales de preparación de péptidos. › sobre soportes.
- C07K7/64 C07K […] › C07K 7/00 Péptidos con 5 a 20 aminoácidos en una secuencia totalmente determinada; Sus derivados. › Péptidos cíclicos que contienen solamente enlaces peptídicos normales.
PDF original: ES-2550856_T3.pdf
Fragmento de la descripción:
Peptidomiméticos en horquilla β fijados sobre una plantilla, con actividad antagonista con respecto a CXCR4
La presente invención da a conocer peptidomiméticos de horquilla β fijados sobre plantilla que incorporan cadenas fijadas sobre plantilla de 14 residuos de α aminoácidos que dependiendo de sus posiciones sobre las cadenas son Gly, NmeGly, Pro o Pip, o de ciertos tipos, tal como se definen más adelante. Estos miméticos de horquilla β fijados sobre plantilla tienen actividad antagonista con respecto a CXCR4. Además, la presente invención da a conocer un procedimiento sintético eficaz por el que estos compuestos pueden, en caso deseado, ser realizados en formato paralelo de librería. Estos peptidomiméticos de horquilla β muestran una eficacia mejorada, biodisponibilidad, media vida y de manera más importante una proporción significativamente incrementada entre la actividad antagonista con CXCR4 por una parte y la hemólisis sobre glóbulos rojos y citotoxicidad por otra.
Hasta el momento, las terapias disponibles para el tratamiento de infecciones VIH han llevado a una notable mejora de los síntomas y a la recuperación de la enfermedad en personas infectadas. Si bien la terapia altamente activa antirretrovírica (terapia HAART) que comporta una combinación de transcriptas inversa/inhibidor de proteasa ha mejorado notablemente el tratamiento clínico de individuos con infección de sida o VIH, subsisten todavía varios problemas graves incluyendo la resistencia a múltiples medicamentos, efectos adversos significativos y costes elevados. Son particularmente deseados agentes antiVIH que bloqueen la infección VIH en una etapa previa de la infección, tal como la entrada vírica.
Se ha reconocido recientemente que para una eficiente entrada en las células objetivo, los virus de la inmunodeficiencia humana requieren los receptores de quimioquina CCR5 y CXCR4, así como el receptor primario CD4 (N. Levy, Engl. J. Med., 335, 29, 1528-1530) . De acuerdo con ello, un agente que pueda bloquear los receptores de quimioquina CXCR4 deben impedir infecciones en individuos sanos y reducir o detener el avance vírico en pacientes infectados (Science, 1997, 275, 1261-1264) .
Entre los diferentes tipos de inhibidores de CXCR4 (M. Schwarz, T. N. C. Wells, A.E.I. Proudfoot, Receptors and Channels, 2001, 7, 417-428) , una clase emergente se basa en análogos de péptidos catiónicos de aparición natural derivados de polifemusina II que tienen una estructura de hoja β antiparalela y una horquilla β que se mantiene por dos puentes de bisulfuro (H. Nakashima, M. Masuda, T. Murakami, Y. Koyanagi, A. Matsumoto, N. Fuji, N.Yamamoto, Antimicrobial Agents and Chemoth. 1992, 36, 1249-1255; H. Tamamura, M. Kuroda, M. Masuda, A. Otaka, S. Funakoshi, H. Nakashima, N. Yamamoto, M. Waki, A. Matsumotu, J.M. Lancelin, D. Kohda, S. Tate, F. Inagaki, N. Fujii, Biochim. Biophys. Acta 1993, 209, 1163; WO 95/10534 A1) .
Las síntesis de análogos estructurales y estudios estructurales por espectroscopia de resonancia magnética nuclear (RMN) han mostrado que los péptidos catiónicos adoptan conformaciones de horquilla β bien definidas debido al efecto limitativo del uno o dos puentes de bisulfuro (H. Tamamura, M. Sugioka, Y. Odagaki, A. Omagari, Y. Kahn, S. Oishi, H. Nakashima, N. Yamamoto, S.C. Peiper, N. Hamanaka, A. Otaka, N. Fujii, Bioorg. Med. Chem. Lett. 2001, 359-362) . Estos resultados muestran que la estructura de horquilla β juega un importante papel en la actividad antagonista con respecto a CXCR4.
Otros estudios estructurales adicionales han indicado también que la actividad antagonista puede verse influida también por la modulación de la estructura amfifílica y el farmacoforo (H. Tamamura, A. Omagari, K. Hiramatsu, K. Gotoh, T. Kanamoto, Y. Xu, E. Kodama, M. Matsuoka, T. Hattori, N. Yamamoto, H. Nakashima, A. Otaka, N. Fujii, Bioorg. Med. Chem. Lett. 2001, 11, 1897-1902; H. Tamamura, A. Omagari, K. Hiramatsu, S. Oishi, H. Habashita, T. Kanamoto, K. Gotoh, N. Yamamoto, H. Nakashima, A. Otaka N. Fujii, Bioorg. Med.Chem. 2002, 10, 1417-1426; H. Tamamura, K. Hiramatsu, K. Miyamoto, A. Omagari, S. Oishi, H. Nakashima, N. Yamamoto, Y. Kuroda, T. Nakagawa, A. Otaki, N. Fujii, Bioorg. Med. Chem. Letters 2002, 12, 923-928) .
Un tema clave en el diseño de péptidos con efecto antagonista sobre CXCR4 es la selectividad. Los análogos derivados de polifemusina II ejercen todavía citotoxicidad a pesar de las mejoras conseguidas (K. Matsuzaki, M. Fukui, N. Fujii, K. Miyajima, Biochim. Biophys. Acta 1991, 259, 1070; A. Otaka, H. Tamamura, Y. Terakawa, M. Masuda, T. Koide, T. Murakami, H. Nakashima, K. Matsuzaki, K. Miyajima, T. Ibuka, M. Waki, A. Matsumoto, N. Yamamoto, N. Fujii Biol. Pharm. Bull. 1994, 17, 1669 y referencias citadas anteriormente) .
Esta actividad citotóxica obvia esencialmente su utilización in vivo, y representa una serie de desventajas en aplicaciones clínicas. Antes de que se pueda considerar la utilización intravenosa, la toxicidad general, actividad de unión a proteínas en el suero sanguíneo, y también la estabilidad como proteasa son temas graves que deben ser enfocados adecuadamente.
Recientemente se ha demostrado que el receptor de CXCR4 no está involucrado solamente en la entrada de VIH sino también en la actividad quimiotáctica de células de cáncer, tales como metastasis de cáncer de seno o metastasis de cáncer de ovarios (A. Muller, B. Homey, H. Soto, N. Ge, D. Catron, M.E. Buchanan, T. Mc Clanahan,
E. Murphey, W. Yuan, S.N. Wagner, J. Luis Barrera, A. Mohar, E. Verastegui, A. Zlotnik, Nature 2001, 50, 410, J. M.
Hall, K. S. Korach, Molecular Endocrinology, 2003, 1-47) , Non-Hodgin’s Lymphoma (F. Bertolini, C. DellÀgnola, P. Manusco, C. Rabascio, A. Burlini, S. Monestiroli, A. Gobbi, G. Pruneri, G. Martinelli, Cancer Research 2002, 62, 3106-3112) , or lung cancer (T.Kijima, G. Maulik, P. C. Ma, E. V. Tibaldi, R:E. Turner, B. Rollins, M. Sattler, B.E. Johnson, R. Salgia, Cancer Research 2002, 62, 6304-6311) , melanoma, cáncer de próstata, cáncer de riñón, neuroblastomia, cáncer de páncreas, mieloma múltiple, leucemia linfocítica crónica (H. Tamamura y otros Febs Letters 2003, 550 79-83, cited ref.) . El bloqueo de la actividad quimitáctica con un inhibidor de CXCR4 debe detener la migración de células de cáncer.
El receptor de CXCR4 ha sido implicado también en el crecimiento y proliferación de tumores. Se ha demostrado que la activación del receptor de CXCR4 era crítica para el crecimiento de tumores malignos tanto neuronales como gliales y tumores de pulmón de células pequeñas. Además, la administración sistémica del antagonista AMD3100 de CXCR4 inhibe el crecimiento de glioblastoma intracraneal e injertos de meduloblastoma al incrementar la apoptosis y disminuir la proliferación de células tumorales (Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K, Kieran MW, Luster AD, Segal RA. Proc Natl Acad Sci U S A. 2003 100 (23) :13513-13518, Barbero S, Bonavia R, Bajetto A, Porcile C, Pirani P, Ravetti JL, Zona GL, Spaziante R, Florio, T. Schettini G. Stromal Cancer Res. 2003, 63 (8) : 19691974, Kijima T, Maulik G, Ma PC, Tibaldi EV, Turner RE, Rollins B. Sattler M. Johnson BE, Salgia R. Cancer Res. 2002-; 62 (21) : 6304-6311, Cancer Res. 2002;62 (11) :3106-3112.
El factor derivado de células del estroma 1 (CXCL12/SDF-1) y su receptor CXCR4 están involucrados en el tráfico de células B y progenitores hematopoiéticos. Se ha demostrado que el receptor de CXCR4 juega un importante papel en la liberación de células madre desde el tuétano del hueso a la sangre periférica. El receptor es expresado, por ejemplo, sobre células CD34+, y ha sido implicado en el proceso de la emigración y la instalación de células CD34+. Esta actividad del receptor de CXCR4 puede ser muy importante para recogidas eficientes de aferesis de células madre de sangre periférica. Las células de sangre periférica autólogas proporcionan una recuperación hematopoiética rápida y mantenida, a continuación del autotransplante, después de la administración de quimioterapia o radioterapia en grandes dosis en pacientes con enfermedades malignas hematológicas y tumores sólidos. (WC. Liles y otros, Blood 2003, 102, 2728-2730) .
Existen pruebas crecientes que sugieren que las quimioquinas, en general y la interacción entre el factor alfa quimioatractivo, derivado de CXCL12/células del estroma y su receptor CXCR4 en particular, juegan un papel determinante en la angiogénesis. Las quimioquinas inducen angiogénesis directamente al unir sus receptores sobre células endoteliales o indirectamente promoviendo infiltrados celulares inflamatorios, que facilitan otros estímulos angiogénicos. Se ha demostrado que una... [Seguir leyendo]
Reivindicaciones:
1. Compuesto de fórmula en la que es un residuo de dipéptido constituido por dos elementos constructivos de aminoácidos diferentes, siendo el dipéptido DPro-LPro; y Z es una cadena constituida por 12 residuos de α-aminoácidos, en la que
-el residuo P1 es el residuo de Tyr;
-el residuo P2 es el residuo de Arg o Gly; -el residuo P3 es el residuo de Cit; -el residuo P4 es el residuo de Val, Phe, Cys, Gly Ile, Thr o Gln; -el residuo P5 es el residuo de Arg; -el residuo P6 es el residuo de DArg o Arg;
-el residuo P7 es el residuo de Arg; -el residuo P8 es el residuo de 2-Nal o Trp; -el residuo P9 es el residuo de Phe, Val, Cys, Gly, Ile, Thr o Gln; -el residuo P10 es el residuo de Tyr; -el residuo P11 es el residuo de Cit o Gly;
-el residuo P12 es el residuo de Lys;
Cys en P4 y P9 forman un puente de bisulfuro en forma libre o en forma de una sal farmacéuticamente aceptable.
3. Compuesto, según la reivindicación 2, que tiene actividad antagonista de CXCR4 y/o actividad anticáncer y/o actividad anti inflamatoria.
5. Composición, según la reivindicación 4, en forma apropiada para administración oral, tópica, transdérmica,
inyección, bucal, transmucosal, pulmonar o inhalación. 40
6. Composición, según la reivindicación 4 ó 5, en forma de tabletas, grageas, cápsulas, soluciones, líquidos, geles, parches, cremas, ungüentos, jarabes, emulsiones, suspensiones, pulverizaciones, nebulizadores o supositorios.
7. Utilización de un compuesto, según la reivindicación 1, para la fabricación de medicamento antagonista de 45 CXCR4.
8. Utilización, según la reivindicación 7, en la que dicho medicamento antagonista de CXCR4 está destinado a su utilización para prevenir infecciones de VIH en individuos sanos, o para hacer más lento y detener el avance vírico en pacientes infectados; o en los que resulta cáncer o es mediado, o en el caso de que una enfermedad
inmunológica es mediada o resulta de actividad del receptor de CXCR4; o para tratar la inmunosupresión; o durante recogidas de células madre de sangre periférica por aferesis.
9. Procedimiento para la fabricación de un compuesto, según la reivindicación 1, que comprende
(a) acoplamiento de un soporte sólido apropiadamente funcionalizado con un derivado apropiadamente N protegido de dicho aminoácido que en el producto final deseado se encuentra en posición 6, 7 u 8, estando igualmente protegido de forma apropiada cualquier grupo funcional que pueda encontrarse presente en dicho derivado de aminoácido N-protegido;
(b) eliminar el grupo N-protector del producto obtenido;
(c) acoplar el producto obtenido de este modo con un derivado apropiadamente N-protegido de dicho aminoácido que en los productos finales deseados se encuentra una posición más próximo al residuo de aminoácido N-terminal, siendo igualmente protegido de forma apropiada cualquier grupo funcional, que pueda encontrarse presente en dicho derivado de aminoácido N-protegido;
(d) eliminar el grupo N-protector obtenido;
(e) repetir las etapas (c) y (d) hasta que el residuo de aminoácido del terminal N ha sido introducido;
(f) acoplamiento del producto obtenido con un compuesto de fórmula general;
en la que es la definida anteriormente y X es un grupo N-protector
(g) eliminando el grupo N-protector del producto obtenido en la etapa (f) o en la etapa (fc) ;
(h) acoplando el producto obtenido de este modo con un derivado apropiadamente N-protegido de dicho aminoácido que en el producto final deseado se encuentra en posición 14, siendo igualmente protegido de modo apropiado cualquier grupo funcional que puede encontrarse presente en dicho derivado de aminoácido N-protegido;
(i) eliminar el grupo N-protector del producto obtenido de este modo;
(j) acoplar el producto obtenido con un derivado apropiadamente N-protegido de dicho aminoácido que en el producto final deseado se encuentra una posición más alejado de la posición 14, encontrándose igualmente protegido de forma apropiada cualquier grupo funcional que pueda encontrarse presente en dicho derivado de aminoácido N-protegido;
(k) eliminar el grupo N-protector del producto obtenido de este modo;
(l) repetir las etapas (j) y (k) hasta que todos los residuos de aminoácidos han sido introducidos; (m) en caso deseado desproteger selectivamente uno o varios grupos funcionales protegidos presentes en la molécula y sustituir de manera apropiada el grupo o grupos reactivos liberados de este modo:
(n) en caso deseado formar uno, dos o tres enlaces entre hebras entre cadenas laterales de residuos de aminoácidos apropiados en posiciones opuestas de la región de hebra β;
(o) desacoplar el producto obtenido del soporte sólido;
(p) ciclar el producto fraccionado del soporte sólido;
(q) eliminar cualesquiera grupos protectores presentes en grupos funcionales de cualesquiera miembros de la cadena de residuos de aminoácidos y, en caso deseado, cualquier grupo o grupos protectores que pueden encontrarse presentes además en la molécula; y
(r) en caso deseado, convertir el producto obtenido de este modo en una sal farmaceuticamente aceptable
10. Procedimiento para la fabricación de un compuesto, según la reivindicación 1, que comprende (a’) acoplamiento de un soporte sólido apropiadamente funcionalizado con un compuesto de fórmula general
en la que es la definida anteriormente y X es un grupo N-protector (b’) eliminar el grupo N-protector del producto obtenido en la etapa (a’) o en la etapa (a’c) ; (c’) acoplar el producto obtenido de este modo con un derivado apropiadamente N-protegido de dicho aminoácido que en el producto final deseado se encuentra en posición 18, encontrándose igualmente protegido de forma apropiada cualquier grupo funcional que pueda encontrarse presente en dicho derivado de aminoácido N-protegido; (d’) eliminar el grupo N-protector del producto obtenido de este modo; (e’) acoplar el producto obtenido de este modo con un derivado apropiadamente N-protegido de dicho aminoácido que en el producto final se encuentra una posición más allá de la posición 12, encontrándose igualmente protegido de forma apropiada cualquier grupo funcional que pueda encontrarse presente en dicho derivado de aminoácido N-protegido; (f’) eliminar el grupo N-protector del producto obtenido de este modo; (g’) repetir las etapas (e’) y (f) hasta que sean introducidos todos los residuos de aminoácidos;
(h’) en caso deseado, desproteger selectivamente uno o varios grupos funcionales protegidos presente en la molécula y sustituir de manera apropiada el grupo o grupos reactivos liberados de esta forma; (i’) en caso deseado formar uno, dos o tres enlaces interhebras entre cadenas laterales de residuos de aminoácidos apropiados en posiciones opuestas de la región de la hebra β; (j’) desacoplar el producto obtenido del soporte sólido;
(k’) ciclar el producto fraccionado del sopote sólido; (l’) eliminar cualesquiera grupos protectores presentes en grupos funcionales de cualesquiera miembros de la cadena de residuos de aminoácidos, y en caso deseado, cualquier grupo o grupos protectores que se pueden encontrar adicionalmente presentes en la molécula; y (m’) en caso deseado, convertir el producto obtenido de este modo en una sal farmaceuticamente aceptable o convertir una sal farmaceuticamente aceptable, o inaceptable, obtenida de este modo en el compuesto libre correspondiente de fórmula I o en una sal diferente, farmaceuticamente aceptable.
Patentes similares o relacionadas:
Péptidos de unión beta amiloide y sus usos para el tratamiento y el diagnóstico de la demencia de Alzheimer, del 17 de Junio de 2020, de Priavoid GmbH: Péptido que contiene al menos una secuencia de aminoácidos que se une a especies beta amiloides y en el que la carga negativa del grupo carboxilo presente […]
Péptidos cíclicos de localización en NTCP y sus usos como inhibidores de entrada, del 6 de Mayo de 2020, de Ruprecht-Karls-Universität Heidelberg: Un péptido cíclico de la fórmula general Ia cyclo[(X)m-P-(Y)n] (Ia) en el que P es la secuencia de aminoácidos NPLGFXaaP (SEQ. ID NÚM.: […]
Inmunomoduladores, del 8 de Abril de 2020, de BRISTOL-MYERS SQUIBB COMPANY: Un compuesto de fórmula (I) **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en la que: A se selecciona entre; **(Ver fórmula)** […]
Análogos de compstatina con propiedades farmacocinéticas mejoradas, del 23 de Octubre de 2019, de THE TRUSTEES OF THE UNIVERSITY OF PENNSYLVANIA: Un compuesto que comprende un péptido de compstatina modificado (ICVVQDWGHHRCT (C2-C12 cíclico; SEQ ID NO:1), en donde la modificación comprende un componente […]
Peptidomiméticos en horquilla beta, del 24 de Julio de 2019, de Polyphor AG: Compuesto de fórmula general (I), Ciclo[P1-P2-P3-P4-P5-P6-P7-P8-P9-P10-P11-P12-T1-T2] (I) en la que los elementos individuales T o P están conectados en […]
Usos de bremelanotida en una terapia para la disfunción sexual femenina, del 22 de Febrero de 2019, de PALATIN TECHNOLOGIES, INC.: Una composición que comprende bremelanotida o una sal farmacéuticamente aceptable de la misma para usar en un método para tratar la disfunción sexual femenina en una paciente […]
Peptidomiméticos en horquilla beta, del 29 de Noviembre de 2018, de Polyphor AG: Un compuesto de la fórmula general (I), ciclo[P1-P2-P3-P4-P5-P6-P7-P8-P9-P10-P11-P12-T1-T2] (I) en donde los elementos individuales T o P están conectados […]
Péptido antagonista de la unión entre el CD47 y una proteína perteneciente a la familia de las trombospondinas, del 13 de Septiembre de 2018, de UNIVERSITE DE REIMS CHAMPAGNE-ARDENNE: Péptido antagonista de la unión entre un receptor CD47 y una proteína perteneciente a la familia de las trombospondinas, o TSP, caracterizado porque presenta la siguiente […]