Nuevo procedimiento de preparación de 9-desoxo-9a-aza-9a-homoeritromicina modificada en el C-4'''' del anillo de cladinosa por un grupo epóxido.
Procedimiento de fabricación de un compuesto de fórmula**Fórmula**
que comprende las etapas de:
(i) proteger el grupo hidroxilo en el C-2' 5 y, opcionalmente, el grupo oxima en el C-9 de la 9-(E)-oxima de la eritromicina A, para obtener un compuesto de fórmula**Fórmula**
en la que R1 es un grupo protector de hidroxilo y R2 es hidrógeno o un grupo protector de oxima; (ii) oxidar el compuesto de fórmula (3) en presencia de un agente oxidante, con objeto de obtener un compuesto de fórmula**Fórmula**
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/EP2011/066205.
Solicitante: Novartis Tiergesundheit AG.
Nacionalidad solicitante: Suiza.
Dirección: Werk Rosental, Schwarzwaldallee 215, WRO-1032 4058 Basel SUIZA.
Inventor/es: GARCIA, RAFAEL, MARTORELL,ORIOL, CODONY,ALBERT.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C07H1/00 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07H AZUCARES; SUS DERIVADOS; NUCLEOSIDOS; NUCLEOTIDOS; ACIDOS NUCLEICOS (derivados de ácidos aldónicos o sacáricos C07C, C07D; ácidos aldónicos, ácidos sacáricos C07C 59/105, C07C 59/285; cianohidrinas C07C 255/16; glicales C07D; compuestos de constitución indeterminada C07G; polisacáridos, sus derivados C08B; ADN o ARN concerniente a la ingeniería genética, vectores, p. ej. plásmidos o su aislamiento, preparación o purificación C12N 15/00; industria del azúcar C13). › Procesos para la preparación de derivados de azúcar.
- C07H17/08 C07H […] › C07H 17/00 Compuestos que contienen radicales heterocíclicos unidos directamente a los heteroátomos de los radicales sacárido. › Heterociclos que contienen ocho o más miembros cíclicos, p. ej. eritromicinas.
PDF original: ES-2532455_T3.pdf
Fragmento de la descripción:
Nuevo procedimiento de preparación de 9-desoxo-9a-aza-9a-homoeritromicina modificada en el C-4" del anillo de cladinosa por un grupo epóxido
La presente Invención se refiere a un nuevo procedimiento de preparación de 9-desoxo-9a-aza-9a-homoer¡tromicina 5 modificada en el C-4" del anillo de cladinosa por un grupo epóxido. Este compuesto es, por ejemplo, un Importante Intermedio de la síntesis de tulatromicina.
Antecedentes de la invención
La presente Invención se refiere a un nuevo procedimiento de preparación del compuesto de fórmula
**(Ver fórmula)**El compuesto es adecuado para la introducción de una nueva funcionalidad en la posición C-4" del anillo de cladinosa de la azaeritromicina A, normalmente un tercer grupo básico amino u otra funcionalidad de heteroátomo.
Un objeto adicional de la invención son los nuevos intermedios del procedimiento de la invención. Estos nuevos compuestos pueden aislarse en forma cristalina como sólidos estables con un elevado rendimiento y pureza.
Los macrólidos son una clase bien conocida de antibióticos. La clarltromicina y la azitromicina se han usado durante 15 bastante tiempo para el tratamiento de infecciones respiratorias humanas causadas por diversos patógenos. Más recientemente se ha aprobado la tulatromicina para el tratamiento o la prevención de enfermedades respiratorias bacterianas en el ganado y en cerdos. La tulatromicina es un antibiótico azálldo de 15 miembros de fórmula
**(Ver fórmula)**Los procedimientos de preparación de la tulatromicina, conocidos, por ejemplo, a partir del documento WOOO/3197, 2 del documento EP 98831 o del documento EP 1253153, usan 9-desoxo-9a-aza-9a-homoeritromicina A (azaeritromicina A) como material de partida. Después de la protección del grupo amino 9a y del grupo hidroxilo C-2' con cloroformiato de bencilo, el grupo hidroxilo en la posición 4 es oxidado, y después de la desprotección es convertido en el epóxido de fórmula (1) anterior, que a su vez es convertido en la tulatromicina mediante la reacción del grupo epóxido con n-propilamina.
Los principales inconvenientes de la síntesis conocida son:
(i) El uso del altamente tóxico cloroformiato de bencilo como agente protector, del que se sabe que es un carcinógeno humano por sí mismo o como consecuencia de las impurezas que contaminan habitualmente este reactivo. Por otro lado, está clasificado como un contaminante marino, aumentando así las preocupaciones adicionales por la seguridad, especialmente cuando se manipula a escala industrial.
(¡i) El aislamiento y la purificación de los intermedios del procedimiento y de la tulatromicina requieren una laboriosa cromatografía en columna con objeto de eliminar las impurezas y los productos secundarios formados durante las reacciones.
(iii) Es necesaria una hidrogenación catalítica durante tres días para eliminar los grupos protectores introducidos después de la etapa de oxidación. Esta hidrogenación se lleva a cabo en presencia de metales tóxicos de clase 1 (es decir: paladio) que necesitan ser adecuadamente eliminados hasta unos niveles muy bajos y no deben llegar hasta la tulatromicina.
(iv) La inestabilidad de los azálidos, en particular si el nitrógeno de la posición 9a no está metilado, provoca la degradación del esqueleto de 15 miembros del azálido, como bien se sabe y está documentado en la literatura.
Por lo tanto, existe la necesidad de un procedimiento mejorado de producción de tulatromicina que sea factible a escala industrial. Ahora se ha encontrado sorprendentemente una síntesis suave y aumentable de escala para la tulatromicina y su precursor de la anterior fórmula (1) partiendo de la 9-(E)-oxima de la eritromicina A en lugar de la azaeritromicina A.
Sumario de la invención
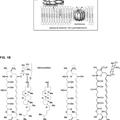
La presente invención se refiere en un aspecto a un nuevo procedimiento de producción de (2R,3S,4R,5R,8R, 1R, 11R, 12S, 13S, 14R)-2-etil-3,4,1-trihidroxi-13-[[(3S,4S,6R,8R)-8-metoxi-4,8-dimetil-1,5- dioxaspiro[2.5]oct-6-il]oxi]-3,5,8,1,12,14-hexametil-11-[[3,4,6-tridesox¡-3-(dimet¡lamino)-a-D-x¡lohexopirano-sil]oxi]-1- oxa-6-azaciclopenatdecano-15-ona de fórmula (1) anterior que comprende las etapas de:
(i) proteger el grupo hidroxilo en el C-21 y, opcionalmente, el grupo oxima en el C-9 de la 9-(E)-oxima de la eritromicina A, para obtener un compuesto de fórmula
en la que Ri es un grupo protector de hidroxilo y R2 es hidrógeno o un grupo protector de oxima;.
(ii) oxidar el compuesto de fórmula (3) en presencia de un agente oxidante con objeto de obtener un compuesto de fórmula
r2o.
N
**(Ver fórmula)** **(Ver fórmula)** **(Ver fórmula)** **(Ver fórmula)**en la que Ri y R2 son como se han definido anteriormente;
(iii) convertir el compuesto de fórmula (4) en el epóxido correspondiente de fórmula
**(Ver fórmula)**en la que R1 y R2 son como se han definido anteriormente;
(iv) retirar el grupo protector R1 y opcionalmente el R2 del compuesto de fórmula (5) para producir el compuesto de fórmula
**(Ver fórmula)**(6);
(v) someter la oxima de fórmula (6) a una reordenación de Beckmann con objeto de obtener el 6,9-imino éter correspondiente de fórmula
**(Ver fórmula)**y
(vi) reducir el ¡mino éter de fórmula (7) para obtener el epóxido de fórmula (1).
De acuerdo con una forma de realización adicional de la invención, en primer lugar se prepara el epóxido de fórmula (1) de acuerdo con el procedimiento como se ha descrito anteriormente, y después se convierte en la tulatromlclna de acuerdo con un procedimiento conocido per se, por ejemplo, a partir del documento EP 98831 o del documento 1 EP 1253153.
Algunas formas de realización de la invención adicionales conciernen a los nuevos Intermedios de fórmulas (4), (5), (6) y (7).
Descripción detallada de la invención
La etapa (I) de la síntesis concierne a la protección selectiva del grupo hldroxl en 2' y, opcionalmente, del grupo 15 hldroxllo correspondiente al grupo oxima en la posición 9, mediante el uso de un grupo protector adecuado. Algunos grupos protectores hidroxilo adecuados son, por ejemplo, los mencionados en T. W. Greene y P. G. M. Wuts "Protective Groups in Organic Synthesis", John Wiley & Sons, Nueva York, 1991, junto con grupos protectores hidroxilo familiares para los expertos en la técnica. Preferiblemente, los grupos protectores hidroxilo en la posición 2' (R-i) y en 9 (R2) son cada uno alcanoílo C2-C4, más preferiblemente acetllo o proplonilo, y en particular acetilo.
Consecuentemente, Ri es preferiblemente alcanoílo C2-C4, más preferiblemente acetilo o proplonilo, y en particular acetllo. R2 es preferiblemente hidrógeno (H) o alcanoílo C2-C4, más preferiblemente H o acetllo o propionllo, y en particular H o acetilo.
El material de partida de 9-(E)-oxima de la eritromicina A está disponible comercialmente o puede prepararse fácilmente de acuerdo con procedimientos sintéticos conocidos per se, por ejemplo, a partir de Tetrahedron Letters,
197, pág.157.
El grupo hidroxilo en la posición C-2' y el grupo hidroxilo de la oxlma pueden ser protegidos, por ejemplo, mediante el tratamiento con un ácido orgánico adecuado o un derivado del mismo, por ejemplo, con un anhídrido de ácido o con un haluro de ácido, en un disolvente aprótico polar, aproximadamente a la temperatura ambiente. La introducción de los grupos acetilo preferidos se consigue, por ejemplo, mediante el tratamiento de la 9-(E)-oxima de la eritromicina A con aproximadamente 2,5 equivalentes de anhídrido acético en un disolvente orgánico aprótico polar, por ejemplo, diclorometano, a la temperatura ambiente, de acuerdo con el procedimiento descrito por J. Berge y col., en el documento W24/39822. Un disolvente alternativo para esta reacción es, por ejemplo, una cetona tal como acetona o metil isobutil cetona, un éter tal como metil terc-butil éter, o un éster del ácido acético tal como acetato de etilo, acetato de isopropilo o acetato de isobutilo. De acuerdo con una forma de realización preferida, tanto el grupo hidroxilo en la posición C-2' como el grupo hidroxilo de la oxima en la posición 9 son protegidos mediante una acilación, en particular, mediante una acetilación, esto es, Ri y R2 son ambos alcanoílo, en particular acetilo.
Los compuestos de fórmula (3) pueden ser procesados adicionalmente sin aislamiento, o pueden ser cristalizados en un disolvente o mezcla de disolventes apropiados, recogerse mediante filtración y procesarse adicionalmente con o sin secado.
La oxidación de los compuestos de fórmula (3) a las correspondientes cetonas de fórmula (4) de acuerdo con la etapa (ii) se lleva a cabo, por ejemplo,... [Seguir leyendo]
Reivindicaciones:
1. Procedimiento de fabricación de un compuesto de fórmula
**(Ver fórmula)**que comprende las etapas de:
(i) proteger el grupo hidroxilo en el C-2' y, opcionalmente, el grupo oxima en el C-9 de la 9-(E)-oxima de la
eritromicina A, para obtener un compuesto de fórmula
**(Ver fórmula)**en la que Ri es un grupo protector de hidroxilo y R2 es hidrógeno o un grupo protector de oxima;
(ii) oxidar el compuesto de fórmula (3) en presencia de un agente oxidante, con objeto de obtener un compuesto 1 de fórmula
**(Ver fórmula)**en la que Ri y R2 son como se han definido anteriormente;
(iii) convertir el compuesto de fórmula (4) en el epóxido correspondiente de fórmula
**(Ver fórmula)**en la que R1 y R2 son cada uno como se ha definido anteriormente;
(iv) retirar el grupo protector R1 y opcionalmente el R2 del compuesto de fórmula (5) para producir el compuesto de fórmula
HO.
N
**(Ver fórmula)**(6);
(v) someter la oxima de fórmula (6) a una reordenación de Beckmann con objeto de obtener el 6,9-imino éter correspondiente de fórmula
**(Ver fórmula)**y
(vi) reducir el ¡mino éter de fórmula (7) para obtener el epóxldo de fórmula (1).
2. El procedimiento de acuerdo con la reivindicación 1, en el que la oxidación del compuesto de fórmula (3) en la etapa (¡I) se realiza con dlmetllsulfóxido, que es activado por anhídrido trifluoroacético, cloruro de oxalilo, un ácido polifosfórico, un complejo de plndlna-SCb o anhídrido acético, en particular por anhídrido trifluoroacético.
3. El procedimiento de la reivindicación 1 o 2, en el que Ri y R2 en las fórmulas (3) y (4) son ambos alcanoílo C2-C4,
en particular acetilo.
4. El compuesto de fórmula
R2<X
**(Ver fórmula)**ch2ch3
ch3
ch3
(4),
en la que Ri es alcanoílo C2-C4, en particular acetilo, y R2es H o alcanoilo C2-C4, preferiblemente alcanoilo C2-C4, y en particular acetilo.
5. El procedimiento de una cualquiera de las reivindicaciones 1 a 3, en el que la preparación del epóxido de fórmula
(5) de acuerdo con la etapa (iii) comprende la reacción del grupo carbonilo en la posición C-4" con un ión metiluro de sulfonio.
6. El compuesto de fórmula
r2o
**(Ver fórmula)**(5);
en la que R1 es alcanoílo C2-C4, en particular acetilo, y R2 es H o alcanoílo C2-C4, en particular H.
7. El procedimiento de una cualquiera de las reivindicaciones 1 a 3 o 5, en el que la eliminación del grupo protector de R1 y opcionalmente de R2 se realiza mediante un tratamiento con un alcanol C1-C4, en particular con metanol.
8. El compuesto de fórmula
**(Ver fórmula)**(6).
9. El procedimiento de una cualquiera de las reivindicaciones 1 a 3, 5 o 7, en el que la preparación del compuesto de fórmula (7) de acuerdo con la etapa (v) se realiza mediante la reacción de la oxima de fórmula (6) con un derivado
de un ácido sulfónlco, en particular con cloruro de p-toluensulfonllo.
1. El compuesto de fórmula
**(Ver fórmula)**11. El procedimiento de una cualquiera de las reivindicaciones 1 a 3, 5, 7 o 9, en el que la reducción del imino éter de fórmula (7) de acuerdo con la etapa (vi) se realiza con un complejo de un hidruro metálico como agente reductor.
12. Un procedimiento de elaboración de tulatromicina, que comprende hacer reaccionar el compuesto de fórmula (1)
obtenido de acuerdo con el procedimiento de una cualquiera de las reivindicaciones 1 a 3, 5, 7, 9 u 11, con n- propilamina.
Patentes similares o relacionadas:
Polimorfos de 20,23-piperidinil-5-O-micaminosiltilonolida, del 20 de Mayo de 2020, de INTERVET INTERNATIONAL B.V: Una forma cristalina de 20,23-dipiperidinil-5-O-micaminosil-tilonolida que tiene al menos una de las siguientes características: un espectro FT-Raman que comprende […]
Derivados de azitromicina con propiedades de potenciación de la barrera epitelial, del 13 de Mayo de 2020, de EpiEndo Pharmaceuticals ehf: Un compuesto de acuerdo con la fórmula (I) **(Ver fórmula)** En donde R1 es OH; R2 es de acuerdo con la fórmula (III) **(Ver […]
Derivados de avermectina B1 que tienen un sustituyente alcoximetilo en la posición 4'''' o 4'', del 17 de Febrero de 2020, de Boehringer Ingelheim Animal Health USA Inc: Compuesto de fórmula**Fórmula** en la que n es 0 o 1; A-B es -CH=CH; R1 es sec-butilo o isopropilo; R2 es CH2CH3, p-ClC6H4, CH2(CH2)2N3 […]
Levoisovalerilespiramicina III y preparaciones, métodos de preparación y utilizaciones de la misma, del 1 de Enero de 2020, de Shenyang Fuyang Pharmaceutical Technology Co., Ltd: Compuesto cristalino de levoisovalerilespiramicina III, en el que, la fórmula estructural química de la levoisovalerilespiramicina III se representa como la fórmula […]
 Derivado de anfotericina b con toxicidad reducida, del 25 de Septiembre de 2019, de THE BOARD OF TRUSTEES OF THE UNIVERSITY OF ILLINOIS: C2'epiAmB, representado por**Fórmula**
o una sal farmacéuticamente aceptable del mismo.
Derivado de anfotericina b con toxicidad reducida, del 25 de Septiembre de 2019, de THE BOARD OF TRUSTEES OF THE UNIVERSITY OF ILLINOIS: C2'epiAmB, representado por**Fórmula**
o una sal farmacéuticamente aceptable del mismo.
Forma cristalina de levoisovalerilespiramicina II y preparaciones, procedimientos de preparación y utilizaciones de la misma, del 7 de Agosto de 2019, de Shenyang Fuyang Pharmaceutical Technology Co., Ltd: Compuesto de levoisovalerilespiramicina II, en el que, la fórmula estructural química de la levoisovalerilespiramicina II se representa como la fórmula (II), en condiciones […]
Derivados de la anfotericina B con índice terapéutico mejorado, del 20 de Septiembre de 2018, de The Board of Trustees of the University of Illionis: Un compuesto, o una sal farmacéuticamente aceptable del mismo, el compuesto que se selecciona de**Fórmula**
Procedimientos para tratar enfermedades gastrointestinales, del 11 de Abril de 2018, de Cempra Pharmaceuticals, Inc: Una cantidad terapéuticamente efectiva de un compuesto de fórmula **Fórmula** o una sal farmacéuticamente aceptable del mismo, donde: R10 es hidrógeno […]