Nueva enzima altamente funcional que tiene especificidad de sustrato modificada.
Una proteína que tiene actividad de α-galactosidasa descrita en (a) o (b):
(a) una proteína que contiene una secuencia de aminoácidos formada por el aminoácido en la posición 18 al aminoácido en la posición 411 incluidos en una secuencia de aminoácidos que se muestra en la secuencia Nº 2 en la cual el aminoácido en la posición 188 está sustituido con ácido glutámico o ácido aspártico y el aminoácido en la posición 191 está sustituido con leucina, valina, isoleucina, fenilalanina o metionina; o
(b) una proteína que contiene una secuencia de aminoácidos descrita en (a), en la cual de uno a diez aminoácidos distintos de los aminoácidos localizados en los sitios de sustitución están sustituidos, en la que los aminoácidos en las posiciones 28 a 31, los aminoácidos en las posiciones 77 a 81, los aminoácidos en las posiciones 117 a 127, los aminoácidos en las posiciones 150 a 158, el aminoácido en la posición 192, los aminoácidos en las posiciones 209 a 220, los aminoácidos en las posiciones 242 a 254 y los aminoácidos en las posiciones 45, 124, 177, 201, 350, 359 y 385 no están mutados.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/JP2006/323509.
Solicitante: Tokyo Metropolitan Institute of Medical Science.
Nacionalidad solicitante: Japón.
Dirección: 1-6, Kamikitazawa 2-chome, Setagaya-ku Tokyo JAPON.
Inventor/es: SAKURABA,HITOSHI, TAJIMA,YOUICHI, AIKAWA,SEIICHI, AIKAWA,FUMIKO, ITO,MAI.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A61K38/00 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › Preparaciones medicinales que contienen péptidos (péptidos que contienen ciclos beta-lactama A61K 31/00; dipéptidos cíclicos que no tienen en su molécula ningún otro enlace peptídico más que los que forman su ciclo, p. ej. piperazina 2,5-dionas, A61K 31/00; péptidos basados en la ergolina A61K 31/48; que contienen compuestos macromoleculares que tienen unidades aminoácido repartidas estadísticamente A61K 31/74; preparaciones medicinales que contienen antígenos o anticuerpos A61K 39/00; preparaciones medicinales caracterizadas por los ingredientes no activos, p. ej. péptidos como soportes de fármacos, A61K 47/00).
- C12N9/24 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12N MICROORGANISMOS O ENZIMAS; COMPOSICIONES QUE LOS CONTIENEN; PROPAGACION, CULTIVO O CONSERVACION DE MICROORGANISMOS; TECNICAS DE MUTACION O DE INGENIERIA GENETICA; MEDIOS DE CULTIVO (medios para ensayos microbiológicos C12Q 1/00). › C12N 9/00 Enzimas, p. ej. ligasas (6.); Proenzimas; Composiciones que las contienen (preparaciones para la limpieza de los dientes que contienen enzimas A61K 8/66, A61Q 11/00; preparaciones de uso médico que contienen enzimas A61K 38/43; composiciones detergentes que contienen enzimas C11D ); Procesos para preparar, activar, inhibir, separar o purificar enzimas. › actúan sobre compuestos glicosílicos (3.2).
- C12N9/40 C12N 9/00 […] › actúan sobre los enlaces alfa-galactosa-glicósido, p. ej. alfa-galactosidasa.
PDF original: ES-2547726_T3.pdf


Fragmento de la descripción:
Nueva enzima altamente funcional que tiene especificidad de sustrato modificada Campo técnico
La presente invención se refiere a una proteína recombinante que tiene actividad de a-galactosidasa.
Técnica antecedente
Para la deficiencia enzimática hereditaria, para la que no se conocen tratamientos radicales hasta la fecha, se ha estado desarrollando terapia de sustitución enzimática en la que se produce una enzima por ingeniería genética y a continuación se administra en un vaso sanguíneo por goteo intravenoso o similar. Como un ejemplo de deficiencia enzimática hereditaria cuya prevalencia es relativamente elevada y que se indica como una enfermedad especificada (enfermedad intratable), se conoce bien la enfermedad de Fabry (deficiencia de a-galactosidasa hereditaria, que es una de un grupo de enfermedades genéticas y también se denomina enfermedad lisosomal) (véase Kenneth J. Dean y col., Fabry Disease, "Practical Enzymology of the Sphingolipidoses", EE.UU., Aln R. Liss, Inc., 1997, páginas 173-216).
La enfermedad de Fabry es un trastorno metabólico de glicolípidos que se desarrolla como sigue a continuación: Como resultado de una disminución de la actividad de una enzima llamada "a-galactosidasa", que es una de las enzimas presentes en un lisosoma, que es uno de los orgánulos intracelulares humanos, y de la deficiencia de la enzima, el glicolípido denominado globotriaosilceramida (también denominado trihexósido de ceramida), que es un sustrato in vivo de la enzima, no se descompone ni se acumula en el organismo (por ejemplo, vasos sanguíneos, pieles, córnea, nervios, riñones y corazón).
Dado que el gen que codifica la a-galactosidasa se encuentra en el cromosoma X, esta enfermedad tiene un modo de herencia por el cromosoma X. Por lo tanto, en esta enfermedad, una característica clínica definitiva se observa principalmente en hemicigoto en machos. Se cree que la "enfermedad de Fabry clásica", que tiene curso clínico habitual, se desarrolla en aproximadamente uno de cada 40.000 niños varones. Síntomas tales como dolor en la mano y en el pie, hipohidrosis, angioqueratoma, y opacidad de la córnea aparecen durante el período juvenil y la adolescencia; estos síntomas evolucionan y entonces provocan daño orgánico sistémico tal como insuficiencia renal, insuficiencia cardiaca, y trastorno cerebrovascular durante la madurescencia y a partir de ese momento; y éstos se convierten en la causa de muerte. Además, una enfermedad que no adquiere tal curso clínico habitual como la "enfermedad de Fabry clásica" y que se desarrolla tarde y adquiere un curso relativamente moderado, es una "variante de la enfermedad de Fabry". En pacientes que tienen este tipo de enfermedad, se observa actividad de a-galactosidasa residual aunque esta es baja. Como una variante de la enfermedad de Fabry, por ejemplo, se conoce la "enfermedad de Fabry cardiaca". La acumulación de glicolípido mencionada anteriormente se produce principalmente en el corazón. Por lo tanto, se produce hipertrofia cardiaca y se provocan trastornos tales como insuficiencia cardiaca y arritmia. Por otro lado, en pacientes heterocigotos hembra de Fabry, se observan diversos tipos de características clínicas de acuerdo con las características del cromosoma X. De forma específica, existen casos que varían de casos graves que son similares a los de los hemicigotos en machos a casos en los que no se observa básicamente ningún síntoma. Sin embargo, de acuerdo con investigación reciente, ha quedado claro que la mayoría de los pacientes heterocigotos hembra de Fabry desarrollan algunos síntomas cuando tienen mayor edad. Existe un punto de vista según el cual no se deberían tratar como "vehículos" sino como "pacientes".
Recientemente, también se ha establecido terapia de sustitución enzimática para la enfermedad de Fabry, y una a- galactosidasa humana recombinante producida en una célula derivada de mamíferos se ha usado ampliamente como un principio activo de un agente terapéutico de la enfermedad de Fabry en la terapia anterior (véase Eng CM y col., Am J Hum Genet, 68: 711-722 (2001); Eng CM y col., N Engl J Med, 345: 9-16 (2001); y Schiffmann R y col., Proc Nati Acad Sci U.S.A, 97: 365-370 (2000)).
Además, también se ha propuesto un procedimiento en el que una a-galactosidasa humana recombinante, producida usando una célula (por ejemplo, levadura) diferente a una célula animal como un huésped, se usa para el tratamiento médico (terapia de sustitución enzimática) de la enfermedad de Fabry (véase la Publicación de Solicitud de Patente Japonesa Sin examinar N° 2002-369692), un procedimiento terapéutico génico en el que una enzima se sustituye mediante la introducción de un gen que codifica a-galactosidasa humana en una célula de un tejido afectado para expresar el gen (véase la Publicación de Solicitud de Patente Japonesa Sin examinar N° (Traducción de la Solicitud de PCT) N° 2002-522509), y similares.
Divulgación de la invención
Sin embargo, dado que a menudo se administra un agente enzimático existente usado en sustitución enzimática para el tratamiento de la enfermedad de Fabry a pacientes que originalmente no tienen una enzima (a-galactosidasa humana), la enzima contenida en el agente terapéutico se reconoce como una sustancia extraña en muchos pacientes administrados con el agente enzimático, y por lo tanto, se produce un anticuerpo. Como resultado, los efectos secundarios adversos, principalmente, las reacciones alérgicas se expresan con una frecuencia elevada. Esto se produce del mismo modo en el caso en el que la enzima se sustituye usando un procedimiento de terapia
génica.
Además, tal agente enzimático usado en sustitución enzimática se administra en los vasos sanguíneos, pero la a-galactosidasa en sí misma es inestable en la sangre. Por consiguiente, en la terapia actual, el agente enzimático se debe administrar frecuentemente (una vez cada dos semanas), y puede ser necesario aumentar la dosificación por administración. Además, la a-galactosidasa humana tiene un número relativamente pequeño de cadenas de azúcar (cadenas de azúcar de tipo N) a la que se puede unir el resto de manosa-6-fosfato (M6P), siendo las cadenas de azúcar necesarias para la a-galactosidasa humana a incorporar en una célula (más específicamente, en un lisosoma en una célula) en un órgano afectado. Por lo tanto, es difícil tomar la a-galactosidasa humana de la sangre e Incorporarla en una célula. En particular, la eficacia de Incorporación en el riñón o el corazón, que es el principal órgano afectado en la enfermedad de Fabry, es baja, y por lo tanto el efecto terapéutico para la nefropatía o cardlopatía es insuficiente. Por consiguiente, con el fin de permitir que una cierta cantidad de enzima se incorpore en una célula diana en la terapia, se requiere una gran cantidad de enzima. En consecuencia, es necesario administrar un agente enzimático usado en sustitución enzimática más frecuentemente y en una cantidad más elevada. Dicha terapia supone una gran carga para los pacientes física, mental y económicamente, y por lo tanto afecta de forma adversa a la "calidad de vida (QOL)".
Por consiguiente, es un objeto de la presente invención proporcionar, como una enzima que se puede usar para sustitución enzimática para la enfermedad de Fabry, una proteína que tiene actividad de a-galactosidasa, que no muestra ningún efecto secundario alérgico adverso, que muestra una estabilidad elevada en sangre (en plasma) y que se puede incorporar fácilmente en una célula de un órgano afectado.
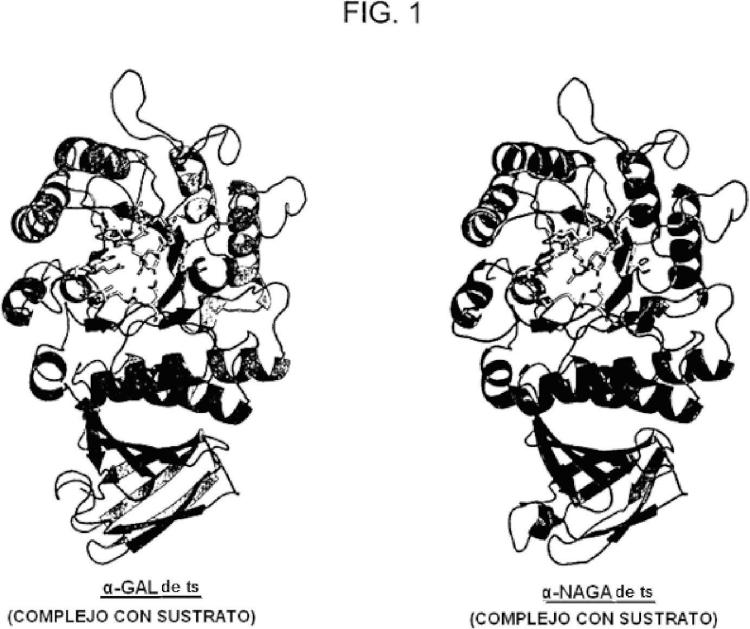
Los presentes inventores dirigieron estudios intensos con el fin de resolver los problemas anteriores. Como resultado, los presentes inventores se centraron en la "a-N-acetilgalactosaminidasa (a-NAGA)", que tiene una especificidad del sustrato diferente de la de la a-galactosidasa pero que tiene una estructura tridimensional muy similar a la de la a-galactosidasa como un conjunto. Los presentes inventores han encontrado que cuando la especificidad del sustrato, específicamente de a-NAGA se convierte con el fin de que tenga actividad de a-galactosidasa cambiando la estructura del sitio activo de a-NAGA usando una técnica de recombinación génica, se puede crear una nueva enzima altamente funcional excelente para tratar la enfermedad de Fabry que se puede usar para solucionar los problemas mencionados anteriormente. Este hallazgo dio como resultado la finalización de la presente invención.
En el contexto de la presente invención se desvela:
(1) Una proteína que tiene actividad adquirida de a-galactosidasa mediante el cambio de la estructura del sitio activo de la a-N-acetilgalactosaminidasa humana de tipo silvestre.
Un ejemplo de la proteína es una proteína que tiene... [Seguir leyendo]
Reivindicaciones:
1. Una proteína que tiene actividad de a-galactosidasa descrita en (a) o (b):
(a) una proteína que contiene una secuencia de aminoácidos formada por el aminoácido en la posición 18 al aminoácido en la posición 411 incluidos en una secuencia de aminoácidos que se muestra en la secuencia N° 2 en la cual el aminoácido en la posición 188 está sustituido con ácido glutámico o ácido aspártico y el aminoácido en la posición 191 está sustituido con leucina, valina, isoleucina, fenilalanina o metionina; o
(b) una proteína que contiene una secuencia de aminoácidos descrita en (a), en la cual de uno a diez aminoácidos distintos de los aminoácidos localizados en los sitios de sustitución están sustituidos, en la que los aminoácidos en las posiciones 28 a 31, los aminoácidos en las posiciones 77 a 81, los aminoácidos en las posiciones 117 a 127, los aminoácidos en las posiciones 150 a 158, el aminoácido en la posición 192, los aminoácidos en las posiciones 209 a 220, los aminoácidos en las posiciones 242 a 254 y los aminoácidos en las posiciones 45, 124, 177, 201, 350, 359 y 385 no están mutados.
2. La proteína de acuerdo con la reivindicación 1, en la que el aminoácido en la posición 188 está sustituido con ácido glutámico y el aminoácido en la posición 191 está sustituido con leucina.
3. Un gen que comprende ADN que codifica la proteína de acuerdo con las reivindicaciones 1 o 2.
4. Un vector recombinante que comprende el gen de acuerdo con la reivindicación 3.
5. Un transformante no humano que comprende el vector recombinante de acuerdo con la reivindicación 4.
6. Un procedimiento de producción de una proteína que tiene actividad de a-galactosidasa, que comprende una etapa de cultivo del transformante de acuerdo con la reivindicación 5 y una etapa de recogida de la proteína que tiene actividad de a-galactosidasa del producto cultivado resultante.
7. Una composición farmacéutica que comprende la proteína de acuerdo con una cualquiera de las reivindicaciones 1 a 2.
8. Un agente terapéutico para la enfermedad de Fabry que, como un principio activo, comprende la composición de acuerdo con la reivindicación 7.
9. Una composición farmacéutica que comprende el gen de acuerdo con la reivindicación 3.
10. Un agente terapéutico génico para la enfermedad de Fabry que, como un principio activo, comprende la composición de acuerdo con la reivindicación 9.
11. Uso de la proteína de acuerdo con cualquiera de las reivindicaciones 1 a 2 o el gen de acuerdo con la reivindicación 3 para la producción de un medicamento para el tratamiento de la enfermedad de Fabry.
12. La proteína de acuerdo con cualquiera de las reivindicaciones 1 a 2 para su uso en el tratamiento de la enfermedad de Fabry.
13. El gen de acuerdo con la reivindicación 3 para su uso en el tratamiento de la enfermedad de Fabry.
Patentes similares o relacionadas:
NANOPARTÍCULAS MULTIFUNCIONALES PARA TERAGNOSIS, del 30 de Julio de 2020, de UNIVERSIDAD DE GRANADA: Nanopartículas multifuncionales para teragnosis. La presente invención se refiere al campo de la medicina, en particular a nanopartículas (NP) […]
Anticuerpos del OPGL, del 15 de Julio de 2020, de AMGEN FREMONT INC.: Un anticuerpo, que comprende una cadena pesada y una cadena ligera, donde: a) la cadena pesada comprende: 1) una secuencia de aminoácidos recogida […]
Formulaciones estables que contienen anticuerpos anti-PCSK9, del 15 de Julio de 2020, de AMGEN INC.: Una formulación estable que comprende un anticuerpo monoclonal que se une específicamente a PCSK9, en donde PCSK9 comprende los aminoácidos de la SEQ ID NO: […]
Polipéptidos de unión a IL-17A, del 15 de Julio de 2020, de AFFIBODY AB: Polipéptido de unión a IL-17A, que comprende un motivo de unión BM a IL-17A, cuyo motivo consiste en una secuencia de aminoácidos seleccionada […]
Modificación de FVIII dirigida al sitio, del 15 de Julio de 2020, de BAYER HEALTHCARE LLC: Un conjugado que tiene actividad procoagulante del factor VIII que comprende un factor VIII polipeptídico funcional con el dominio B eliminado que está mutado […]
Composición a base de hidroxiapatita en polvo para el tratamiento del linfoma B o T, del 1 de Julio de 2020, de URODELIA: Composición para uso como autovacuna antitumoral para el tratamiento de linfomas B o T en un sujeto, que comprende un polvo de hidroxiapatita y/o de […]
Tratamiento de disfunción eréctil y otras indicaciones, del 1 de Julio de 2020, de STRATEGIC SCIENCE & TECHNOLOGIES, LLC: Una composición para su uso en un método de tratamiento de la disfunción sexual en un sujeto, preferiblemente un sujeto humano, comprendiendo la composición: […]
Péptido derivado de GPC3, composición farmacéutica para el tratamiento o la prevención de cáncer usando el mismo, inductor de inmunidad y método para producir células presentadoras de antígeno, del 17 de Junio de 2020, de CYTLIMIC INC: Composición farmacéutica para su uso en el tratamiento o la prevención de cáncer, que comprende un péptido que consiste en una secuencia de aminoácidos […]