Métodos y composiciones para el tratamiento de síndrome de Marfan y trastornos asociados.
Losartán para uso en el tratamiento de síndrome de Marfan.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/US2006/041846.
Solicitante: THE JOHNS HOPKINS UNIVERSITY.
Nacionalidad solicitante: Estados Unidos de América.
Dirección: 3400 N. CHARLES STREET BALTIMORE, MD 21218 ESTADOS UNIDOS DE AMERICA.
Inventor/es: DIETZ,HARRY C, JUDGE,DANIEL P, NEPTUNE,ENID R, COHN,RONALD, HABASHI,JENNIFER.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A61K48/00 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › Preparaciones medicinales que contienen material genético que se introduce en las células del cuerpo vivo para tratar enfermedades genéticas; Terapia génica.
PDF original: ES-2526705_T3.pdf




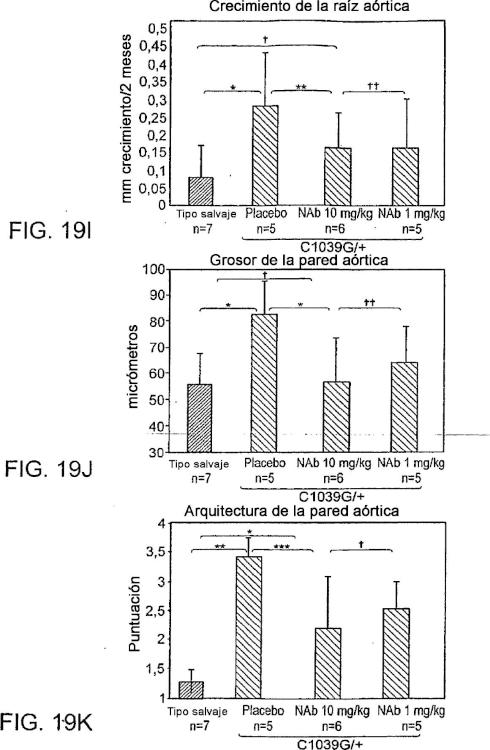
Fragmento de la descripción:
Métodos y composiciones para el tratamiento de síndrome de Marfan y trastornos asociados ANTECEDENTES DE LA INVENCIÓN
El síndrome de Marfan (MFS) es un trastorno sistémico del tejido conjuntivo con herencia dominante autosómlca y una prevalencla de aproximadamente 1 por 5. habitantes (Pyeritz, R.E. y McKusick, V.A. (1979) N Engl J Med. 3, 772-777). El síndrome no muestra preferencia racial, y ambos sexos están afectados igualmente. Se ha estimado que el 25% de los casos se producen debido a mutaciones espontáneas. Aunque esta afección muestra una penetrancia elevada, la norma es la variabilidad clínica interfamiliar marcada (Pyeritz, R.E. et al. (1979) Blrth Defects Orig Artic Ser. 15, 155-178). La falta de un marcador bioquímico o genético específico de la enfermedad, acoplado con la variabilidad en la presentación clínica, ha frustrado el diagnóstico de casos equívocos y ha contribuido probablemente a una subestimación significativa de la prevalencia de la enfermedad.
Las características fundamentales de este trastorno implican los sistemas ocular, esquelético, y cardiovascular. La patología cardiovascular, incluyendo dilatación, disección, y ruptura de la raíz aórtica, dilatación de la arteria pulmonar, cambios en la válvula mixomatosa con insuficiencia de las válvulas mitral y aórtica, y la disfunción miocárdica progresiva, es la causa principal de mortalidad en el MFS. La mayoría de sucesos mortales asociados con MFS sin tratar se produce en la vida adulta temprana. En un estudio potencial de 72 pacientes en 1972, la edad promedio de muerte fue 32 años (Murdoch, J.L. et al. (1972) N Engl J Med. 286, 84-88).
Una reevaluación reciente de la esperanza de vida en el síndrome de Marfan sugirió que el diagnóstico temprano y el manejo médico y quirúrgico refinado han mejorado enormemente esta situación (Silverman, D.l. et al. (1995) Am J Cardiol. 75, 157-16). No obstante, el MFS continúa estando asociado con morbidez significativa, y subgrupos selectos son refractarios a la terapia y continúan mostrando una mortalidad temprana Morse, R.P. et al. (199) Pediatrics. 86, 888-895; Sisk, H.E.; et al. (1983) Am J Cardiol. 52, 353-358). En una revisión de 54 pacientes diagnosticados durante la infancia, Morse et al. dio a conocer que el 89% tuvo patología cardíaca grave, y que la enfermedad cardíaca fue progresiva a pesar del cuidado estándar (el 22% murió durante la niñez, el 16% antes de la edad de 1 año). En la forma más clásica de síndrome de Marfan, se estima que más del 9% de los individuos tendrán un "suceso" cardiovascular durante su vida, definido como la necesidad de reparación quirúrgica profiláctica de la raíz aórtica, o muerte debido a disección aórtica (Gillinov, A.M., et al. (1997) Ann Thorac Surg. 64, 114-1144; discusión 1144-1145; Pyeritz, R.E. (1993) Semln Thorac Cardiovasc Surg. 5, 11-16; Silverman, D.I., et al. (1995) J Am Coll Cardiol. 26, 162-167; Gott, V.L., et al. (1999) N Engl J Med. 34, 137-1313). La morbidez ocular y esquelética se cuantifica de forma menos fácil (Maumenee, I.H. et al. (1981) Trans Am Ophthalmol Soc. 79, 684- 733; Magid, D., et al. (199) AJR Am J Roentgenol. 155, 99-14; Sponseller, P.D., et al. (1995) J Bone Joint Surg Am. 77, 867-876). Aproximadamente el 6% de Individuos con MFS tiene dislocación de la lente, requiriendo a menudo afaquia quirúrgica para el manejo óptimo. El desprendimiento retiniano y el glaucoma pueden provocar una alteración visual devastadora.
La Implicación esquelética es evidente en casi todas las personas con MFS. La deformidad del pecho anterior progresiva o escoliosis puede provocar disfunción cardiopulmonar, y habitualmente requiere corrección quirúrgica. La inestabilidad articular puede provocar incapacidad física y predisponer a artritis prematura. La neumopatía se manifiesta muy habitualmente con neumotorax espontáneo, y se ha identificado en 4-11% de los pacientes con MFS (Wood, J.R, et al. (1984) Thorax. 39, 78-784; Hall, J.R., etal. (1984) Ann Thorac Surg. 37, 5-54). Los hallazgos patológicos Incluyen bulas en el lóbulo superior con o sin enfermedad obstructiva fija difusa de las vías respiratorias que puede ser progresiva y se ha equiparado tradicionalmente con enfisema destructivo (Lipton, R.A., et al. (1971) Am Rev Respir Dis. 14, 924; Domínguez, R., et al. (1987) Pediatr Radiol. 17, 365-369). La mayoría de los pacientes con MFS presenta una deficiencia marcada en la masa muscular esquelética y almacenes de grasa a pesar de la ingesta calórica adecuada y sin signos de mala absorción (Behan, W.M., et al. (23) J Neurol Neurosurg Psychiatry. 74, 633-63 8; H.H., et al. (1973) Neurology. 23, 1257-1268; Gross, M.L., et al. (198) J Neurol Sc¡. 46, 15-112; Joyce, D.A., et al. (1984) Aust N Z J Med. 14, 495-499). Se han observado signos de miopatía muscular esquelética, incluyendo menor fuerza y tono, en un subconjunto de individuos afectados, y puede contribuir a un menor comportamiento funcional, insuficiencia respiratoria, pérdida de alineamiento ocular, y desarrollo alterado del esqueleto, incluyendo cifosis y escoliosis.
Un reto creciente es definir la "nueva" historia natural de MFS ahora que muchos individuos están sobreviviendo a su predisposición para la disección de la raíz aórtica temprana; los fenotipos asociados con el envejecimiento ya apreciados incluyen una predisposición a la disección de la aorta torácica y abdominal descendente. De este modo, a pesar de los avances en nuestra capacidad para incrementar la duración de vida de muchos individuos con MFS, hay una oportunidad más que suficiente para mejorar la calidad de vida de la mayoría de los individuos afectados.
En 1991, un análisis de candidatos posicionales tradicional culminó con la demostración de que la enfermedad produce mutaciones en el gen FBN1 en el cromosoma 15q21.1 que codifica fibrilina-1 (Dietz, H.C., et al. (1991) Nature. 352, 337-339). Desde ese momento, ha habido una generación y caracterización de múltiples modelos de ratón del síndrome de Marfan. Este trabajo ha revolucionado verdaderamente la comprensión de la patogénesis de la enfermedad, y ha conducido a estrategias excitantes para el tratamiento de la patogénesis multisistémica del
síndrome de Marfan.
Muchas de las características del síndrome de Marfan son comunes en la población general, y representan una carga de salud pública tremenda. Éstas incluyen aneurisma aórtica (1-2% de la población en general), prolapso de la válvula mitral (~7%), enfisema (11%), escoliosis (,5%), catarata (3%), artritis (muy habitual), y miopatía (muchas formas genéticas y adquiridas comunes).
En consecuencia, existe la necesidad de métodos y composiciones para el tratamiento del síndrome de Marfan y enfermedades asociadas, trastornos y afecciones, por ejemplo enfermedades, trastornos y afecciones asociados con expresión aberrante de TGF-p.
SUMARIO DE LA INVENCIÓN
La actual invención se basa en el descubrimiento de que los antagonistas de TGF-p tratan efectivamente el síndrome de Marfan y la enfermedad y trastornos relacionados con el síndrome de Marfan, por ejemplo enfermedades, trastornos y afecciones asociados con la expresión aberrante de TGF-p.
En consecuencia, en un aspecto, la invención proporciona métodos para tratar el síndrome de Marfan o una afección clínica asociada con síndrome de Marfan, que comprenden administrar al sujeto una cantidad eficaz de un agente que module la actividad o expresión de TGFp, tratando de ese modo al sujeto.
En una realización relacionada, la enfermedad o trastorno es un aneurisma aórtico, enfermedad de la válvula, enfisema, miopatía, escoliosis, o enfermedad ocular. En una realización específica, la enfermedad ocular se selecciona del grupo que consiste en cataratas, miopía, glaucoma, y desprendimiento retiniano. En una realización relacionada, la enfermedad o trastorno es una enfermedad o trastorno que se refiere a crecimiento, mantenimiento o regeneración muscular, por ejemplo distrofia muscular. En una realización específica, la enfermedad o trastorno es distrofia muscular de Duchenne.
El agente es el antagonista del receptor de angiotensina tipo 1, sal monopotásica de 2-butil-4-cloro-1-[p-(o-1H- tetrazol-5-ilfenil)bencil]imidazol-5-metanol (losartán potásico).
La invención describe métodos para tratar un sujeto que tiene síndrome de Marfan o una afección asociada con Marfan, administrando al sujeto una cantidad eficaz de un agente que modula la actividad o expresión de TGFp, tratando de ese modo al sujeto.
El agente es un antagonista de TGFp, por ejemplo una molécula pequeña, un ácido nucleico, un péptido, un anticuerpo, un scFV, o un fragmento Fab. El anticuerpo es un anticuerpo neutralizante. El agente es un ARNip... [Seguir leyendo]
Reivindicaciones:
1. Losartán para uso en el tratamiento de síndrome de Marfan.
2. Losartán para uso como en la reivindicación 1, en el que se trata un aneurisma aórtico.
3. Losartán para uso como en la reivindicación 1, en el que se trata enfermedad de la válvula.
4. Losartán para uso como en la reivindicación 1, en el que se trata prolapso de la válvula mitral.
5. Losartán para uso como en la reivindicación 1, en el que se trata enfisema.
6. Losartán para uso como en la reivindicación 1, en el que se trata mlopatfa.
7. Losartán para uso como en la reivindicación 1, en el que se trata escoliosis.
8. Losartán para uso como en la reivindicación 1, en el que se trata enfermedad ocular.
9. Losartán para uso como en la reivindicación 1, en el que se trata tablcaclón alveolar.
1. Losartán para uso como en la reivindicación 1, en el que se rescatan la regeneración y arquitectura musculares.
11. Uso de losartán en la preparación de un medicamento para el tratamiento de síndrome de Marfan.
Patentes similares o relacionadas:
Terapia génica para la diabetes, del 8 de Julio de 2020, de UCL Business Ltd: Una molécula de ácido nucleico que comprende una secuencia de nucleótidos que codifica una proteína preproinsulina funcional en donde la secuencia de nucleótidos tiene al menos […]
Composiciones útiles en el tratamiento de la deficiencia de ornitina transcarbamilasa (OTC), del 8 de Julio de 2020, de THE TRUSTEES OF THE UNIVERSITY OF PENNSYLVANIA: Un vector vírico recombinante que comprende una secuencia de ácido nucleico que codifica la proteína ornitina transcarbamilasa humana (hOTC) y secuencias […]
Vacuna de ADN que contiene un epítopo específico de VEGF y/o un epítopo específico de angiopoyetina-2, del 1 de Julio de 2020, de OSAKA UNIVERSITY: Un vector de expresión que codifica un polipéptido del antígeno del núcleo del virus de la hepatitis B quimérico con una inserción para uso en el tratamiento o la profilaxis […]
Composiciones para modular la expresión de SOD-1, del 24 de Junio de 2020, de Biogen MA Inc: Un compuesto antisentido según la siguiente fórmula: mCes Aeo Ges Geo Aes Tds Ads mCds Ads Tds Tds Tds mCds Tds Ads mCeo Aes Geo mCes Te (secuencia […]
Ácido nucleico antisentido, del 24 de Junio de 2020, de NIPPON SHINYAKU CO., LTD.: Un oligómero antisentido de 14 a 32 bases de longitud, que comprende dos unidades de oligómeros conectadas seleccionadas del grupo que consiste […]
Plekhg5 como diana farmacéutica para trastornos neurológicos, del 15 de Junio de 2020, de CONSEJO SUPERIOR DE INVESTIGACIONES CIENTIFICAS (CSIC): Plekhg5 como diana farmacéutica para trastornos neurológicos. La invención hace referencia al uso del gen Plekhg5 como diana farmacológica para el cribado, […]
Método para activar células T auxiliares, del 10 de Junio de 2020, de OTSUKA PHARMACEUTICAL CO., LTD.: Una composición para su uso en el tratamiento o prevención del cáncer mediante la activación de células T auxiliares en un sujeto, en donde dicha composición […]
Ratón nuligénico para Pint que muestra un fenotipo asociado a envejecimiento prematuro, del 10 de Junio de 2020, de REGENERON PHARMACEUTICALS, INC.: Un ratón cuyo genoma comprende una inactivación de un locus del ARN no codificante largo (ARNlnc) Pint endógeno, en donde la inactivación (i) da como resultado que el […]