Método y sistema informático para evaluar anotaciones de clasificación asignadas a secuencias de ADN.
Un método implementado por ordenador para la evaluación de anotaciones de clasificación (112) asignadas a secuencias de ADN (111) almacenadas en una base de datos (11),
comprendiendo el método:
agrupar (S1) las secuencias de ADN (111) basándose en sus anotaciones de clasificación respectivas (112) por especies utilizando sistemas de clasificación establecidos;
determinar para parejas de secuencias de ADN (111) una medida de distancia entre las secuencias de ADN respectivas (111) mediante la alineación (S31) de forma automática de las secuencias de ADN respectivas (111) y determinar la medida de distancia (S41) basada en una puntuación de similitud entre las secuencias de ADN alineadas (111);
determinar una secuencia centroide (S4, S40), teniendo la secuencia centroide (C) una medida global más corta de distancia a las secuencias de ADN (111); y
asignar (S5) a las secuencias de ADN (111) la medida de distancia entre la secuencia de ADN respectiva y la secuencia centroide (C) como un nivel de confianza cuantitativo para la anotación de clasificación asignada a la secuencia de ADN respectiva;
en donde la medida de distancia se determina entre las secuencias de ADN (111) dentro de una especie; y las secuencias centroides se determinan para las secuencias de ADN (111) dentro de cada una de las especies.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/CH2007/000599.
Solicitante: SMARTGENE GMBH.
Nacionalidad solicitante: Suiza.
Dirección: INDUSTRIESTRASSE 16 6300 ZUG SUIZA.
Inventor/es: EMLER,STEFAN, MICHEL,PIERRE-ANDRÉ.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- G06F19/22
- G06F19/24
- G06F19/28
PDF original: ES-2456240_T3.pdf
Fragmento de la descripción:
Método y sistema informático para evaluar anotaciones de clasificación asignadas a secuencias de ADN
Campo de la invención La presente invención se refiere a un método implementado por ordenador y a un sistema informático para la evaluación de anotaciones de clasificación asignadas a secuencias de ADN. Específicamente, la presente invención se refiere a un método implementado por ordenador y a un sistema informático para evaluar anotaciones de clasificación asignadas a secuencias de ADN almacenadas en una base de datos.
Antecedentes de la invención La identificación de formas de vida, basada en secuencias se utiliza cada vez más para fines de diagnóstico. Al ser independiente del crecimiento y del metabolismo, este método ofrece ventajas significativas en términos de velocidad y precisión sobre técnicas convencionales basadas en el cultivo. Genes conservados presentes en todas las bacterias u hongos se amplifican y posteriormente se secuencian utilizando técnicas de secuenciación automatizada. Las secuencias obtenidas se comparan después con referencias en una base de datos. De este modo, incluso materiales aislados raros, inesperados o inusuales se pueden identificar y clasificar rápidamente. El análisis de las secuencias se puede aplicar a todos los genes conservados de todas las formas de vida, particularmente a microorganismos tales como bacterias y hongos. La identificación de microorganismos basada en las secuencias, depende de la comparación de la secuencia característica de la muestra con una base de datos que contiene secuencias de referencia que representan todos los géneros y especies pertinentes. Por tanto, es importante que una base de datos de referencia cumpla los siguientes requisitos:
1) Secuencia exacta: la base de datos contiene secuencias correctas de la diana solicitada, no tiene errores de secuenciación, ni fallos de lectura, no hay huecos artificiales, inserciones, no hay secuencias de vectores.
2) Anotación de clasificación correcta (es decir, denominación de los registros) : las secuencias se anotan correctamente (por ejemplo, los nombres de las especies) y esta información se actualiza con respecto a cambios en la taxonomía.
3) Representativa: la base de datos representa todas las formas de vida pertinentes, por ejemplo, género y especie, incluyendo sus variantes genéticas (intraespecíficas, intragenómicas) .
4) Actualización: las referencias se actualizan con respecto a especies descritas recientemente y a posibles cambios en la taxonomía (véase también 2) .
Actualmente no existe una base de datos de referencia única que cumpla todos estos requisitos. Sin embargo, debido a que la calidad de los resultados de las comparaciones de secuencias depende en gran medida de las referencias disponibles, es crucial que estas bases de datos sean lo más fidedignas posible. En general, los científicos añaden registros a repositorios públicos que tienen una calidad aceptable en términos de contenido de la secuencia y de la anotación (por ejemplo, nombre de la especie) . Sin embargo, hay muchos errores de secuenciación o anotaciones incorrectas en relación con la taxonomía actual. Se producen errores de anotación, por ejemplo, cuando las secuencias se presentan junto con una información incorrecta sobre el organismo o el gen a partir del cual se ha obtenido la secuencia, o con nombres de especies que no están actualizados (por ejemplo, cuando las especies se han clasificado de nuevo taxonómicamente, como es frecuentemente en el caso de bacterias) . Cuando una secuencia de una muestra se busca en una base de datos de referencia, la lista resultante suele mostrar coincidencias correctas e incorrectas indistintamente, dejando en manos de la experiencia del usuario la determinación de qué referencias se identificaron de forma correcta o incorrecta. Por lo tanto, una secuencia correcta con una anotación incorrecta podría aparecer en la parte superior de la lista de coincidencias y, por lo tanto, indicar una identificación errónea de una bacteria, por ejemplo. Debido a que la identificación de agentes patógenos basada en la secuencia se está convirtiendo hoy en día en una parte del trabajo rutinario en los laboratorios de diagnóstico médico, veterinarios e industriales, existe una necesidad de que las búsquedas y las comparaciones de secuencias en bases de datos sean fáciles y fiables, por ejemplo, para la identificación de una especie bacteriana o fúngica o un subtipo de virus, o para hacer cotejar cualquier organismo desconocido con una base de datos de organismos bien caracterizados. En particular, los resultados de la búsqueda y la comparación de la similitud de secuencias se deben proporcionar de forma adecuada con respecto a la experiencia de los técnicos de laboratorio común, que, en general, no tienen experiencia científica o una amplia formación en bioinformática o en taxonomía de organismos (microorganismos) .
El documento de EE.UU. 2007/0083334 describe sistemas y métodos para anotar secuencias biomoleculares. Después de la alineación (es) de secuencias, las secuencias biomoleculares se agrupan con métodos informáticos de acuerdo con un campo de homología progresiva, usando uno o varios algoritmos de agrupamiento. Una secuencia biomolecular se considera que pertenece a una agrupación, si la secuencia comparte una homología de secuencia basada en la alineación, superior a un determinado valor umbral con uno de los miembros de la agrupación. Según el documento de EE.UU. 2007/0083334, la agrupación computacional se puede efectuar usando cualquier programa informático de alineación, disponible comercialmente incluyendo un algoritmo de homología local. Por ejemplo, un
grupo muestra un cierto grado de homología, si los ácidos nucleicos son idénticos entre sí en un 90%.
El documento de EE.UU. 2007/0134692 describe un método y un sistema basado en la alineación para actualizar datos de anotación de un conjunto de sondas. Se generan una o varias agrupaciones mediante la transcripción a través de conjuntos de datos recuperados a partir de una o varias fuentes. Una o varias secuencias de sondas se alinea con una secuencia representativa de una o varias de las agrupaciones. La secuencia representativa se alinea con una secuencia del genoma y la secuencia del genoma se anota con información sobre la ubicación de la sonda. Las secuencias de sondas alineadas se cartografían en la secuencia del genoma, usando la alineación de la secuencia representativa y la secuencia del genoma. Una puntuación se calcula utilizando un número asociado con las secuencias de sondas alineadas y un número asociado con la formación de una ubicación de la sonda asociada con una región de la secuencia del genoma que se corresponde con la secuencia representativa alineada. Registros redundantes se pueden eliminar mediante el método de agrupación. Por ejemplo, si la alineación de transcritos en una agrupación se solapa en >97% en su longitud completa, entonces se determina que son redundantes y solo la secuencia más larga se conserva en la agrupación.
Compendio de la invención Es un objeto de esta invención proporcionar un método implementado por ordenador y un sistema informático para evaluar (y evaluar de nuevo) anotaciones de clasificación que incluyen anotaciones taxonómicas, sistemáticas y/o funcionales, asignadas a secuencias de ADN. En particular, un objeto de la presente invención es proporcionar un método implementado por ordenador y un sistema informático para evaluar cualitativamente las anotaciones de clasificación, de tal manera que anotaciones erróneas y/o dudosas sean patentes para los técnicos de laboratorio que no tienen una amplia experiencia o entrenamiento en bioinformática o en taxonomía de organismos (microorganismos) .
De acuerdo con la presente invención, estos objetos se consiguen especialmente a través de las características de las reivindicaciones independientes. Además, otras realizaciones ventajosas se deducen de las reivindicaciones dependientes y de la descripción.
De acuerdo con la presente invención, los objetos anteriormente mencionados se consiguen en particular, ya que para evaluar las anotaciones de clasificación (incluyendo anotaciones taxonómicas, sistemáticas y/o funcionales) asignadas a secuencias de ADN almacenadas en una base de datos, por ejemplo, una base de datos de referencia, las secuencias de ADN se agrupan, basándose en sus anotaciones de clasificación respectivas, por especies utilizando sistemas de clasificación establecidos para la clasificación taxonómica, sistemática y/o funcional. Posteriormente, para las parejas de secuencias de ADN, se determina en cada caso una medida de distancia entre las secuencias de ADN respectivas. La... [Seguir leyendo]
Reivindicaciones:
1. Un método implementado por ordenador para la evaluación de anotaciones de clasificación (112) asignadas a secuencias de ADN (111) almacenadas en una base de datos (11) , comprendiendo el método:
agrupar (S1) las secuencias de ADN (111) basándose en sus anotaciones de clasificación respectivas (112) por especies utilizando sistemas de clasificación establecidos;
determinar para parejas de secuencias de ADN (111) una medida de distancia entre las secuencias de ADN respectivas (111) mediante la alineación (S31) de forma automática de las secuencias de ADN respectivas (111) y determinar la medida de distancia (S41) basada en una puntuación de similitud entre las secuencias de ADN alineadas (111) ;
determinar una secuencia centroide (S4, S40) , teniendo la secuencia centroide (C) una medida global más corta de distancia a las secuencias de ADN (111) ; y
asignar (S5) a las secuencias de ADN (111) la medida de distancia entre la secuencia de ADN respectiva y la secuencia centroide (C) como un nivel de confianza cuantitativo para la anotación de clasificación asignada a la secuencia de ADN respectiva;
en donde la medida de distancia se determina entre las secuencias de ADN (111) dentro de una especie; y las secuencias centroides se determinan para las secuencias de ADN (111) dentro de cada una de las especies.
2. El método según la reivindicación 1, que comprende además la identificación de valores atípicos dentro de la especie, teniendo los valores atípicos la mayor medida de distancia a la secuencia centroide (C) de la especie respectiva, y marcando las anotaciones (112) como incorrectas para valores atípicos que tienen una medida de distancia más pequeña a una secuencia centroide (C) de otra especie.
3. El método según una de las reivindicaciones 1 o 2, en el que el método comprende además generar (S401) a partir de las puntuaciones de similitud entre las secuencias de ADN (111) un grafo de aristas ponderado, siendo las secuencias de ADN (111) nodos en el grafo, estando conectados los nodos si la puntuación de similitud entre las secuencias de ADN respectivas (111) es positiva, y estando asignada la medida de distancia entre las secuencias de ADN respectivas (111) en cada caso como un peso de la arista; calcular (S402) las densidades de la conectividad local para los nodos en el grafo; y definir agrupaciones (S403) de nodos a través de la agregación progresiva hasta una densidad de conectividad local máxima, siendo la medida de distancia entre las secuencias de ADN (111) asociadas con nodos dentro de una agrupación significativamente más corta que una medida promedio de distancia entre las secuencias de ADN (111) asociadas con los nodos del grafo, en donde preferiblemente el método comprende adicionalmente recibir un valor umbral de agrupación (S405) de un usuario, que responde a mostrar el grafo en una pantalla (33) ; definir las agrupaciones (S403) de nodos aplicando el valor umbral de agrupación como una distancia máxima dentro de la agrupación; y mostrar el grafo (S404) en la pantalla (33) después de aplicar el valor umbral de agrupación.
4. El método según la reivindicación 3, en el que la secuencia de ADN asociada con el nodo que tiene la mayor densidad de conectividad en una agrupación se define como la secuencia centroide (C) de esa agrupación.
5. El método según una de las reivindicaciones 1 a 4, en el que la anotación de clasificación asociada con una secuencia centroide (C) se asigna a secuencias de ADN (111) asociadas con esa secuencia centroide (C) .
6. El método según una de las reivindicaciones 1 a 5, en el que la determinación de la medida de distancia (S41) entre dos secuencias de ADN (111) incluye el cálculo de una puntuación ponderada de similitud dividiendo la puntuación de similitud entre las dos secuencias de ADN (111) entre la menor longitud de las dos secuencias de ADN (111) , y restando la puntuación ponderada de similitud de una.
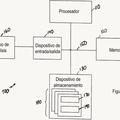
7. Un sistema informático (1) para la evaluación de las anotaciones de clasificación (112) asignadas a secuencias de ADN (111) , comprendiendo el sistema (1) :
una base de datos (11) que comprende una pluralidad de secuencias de ADN (111) ;
un módulo comparador (122) configurado para agrupar las secuencias de ADN (111) basándose en sus anotaciones de clasificación respectivas (112) por especie utilizando sistemas de clasificación establecidos (7) , y para determinar para parejas de las secuencias de ADN (111) una medida de distancia entre las secuencias de ADN respectivas (111) mediante la alineación automática de las secuencias de ADN respectivas (111) y determinar la medida de distancia basada en una puntuación de similitud entre las secuencias de ADN alineadas (111) ;
un detector de centroide (123) configurado para determinar una secuencia centroide (C) , teniendo la secuencia centroide (C) la medida global menor de distancia a las secuencias de ADN (111) ; y
un módulo de valoración (124) configurado para asignar a las secuencias de ADN (111) la medida de distancia entre la secuencia de ADN respectiva y la secuencia centroide (C) como un nivel de confianza cuantitativo para la anota
ción de clasificación asignada a la secuencia de ADN respectiva;
en donde el módulo comparador (122) se configura adicionalmente para determinar la medida de distancia entre las secuencias de ADN (111) dentro de una especie; y el detector de centroide (123) se configura adicionalmente para determinar las secuencias centroides (C) para las secuencias de ADN (111) dentro de cada una de las especies
8. El sistema (1) según la reivindicación 7, que comprende además un detector de errores (125) configurado para identificar valores atípicos dentro de las especies, teniendo los valores atípicos la mayor medida de distancia a la secuencia centroide (C) de la especie respectiva, y para marcar anotaciones (112) como incorrectas para valores atípicos que tienen una medida de distancia más pequeña a una secuencia centroide (C) de otra especie.
9. El sistema (1) según una de las reivindicaciones 7 u 8, en el que el sistema (1) comprende además un generador de grafos (126) configurado para generar a partir de las puntuaciones de similitud entre las secuencias de ADN (111) un grafo de aristas ponderado, siendo las secuencias de ADN (111) nodos en el grafo, estando conectados los nodos si la puntuación de similitud entre las secuencias de ADN respectivas (111) es positiva, y estando asignada en cada caso la medida de la distancia entre las secuencias de ADN respectivas (111) como un peso de arista, para calcular densidades de conectividad locales para los nodos en el grafo, y para definir agrupaciones de nodos a través de la agregación progresiva hasta máximos de densidad de conectividad local, siendo la medida de distancia entre las secuencias de ADN (111) asociada con nodos dentro de una agrupación significativamente más corta que una medida promedio de distancia entre las secuencias de ADN (111) asociadas con los nodos del grafo.
10. El sistema (1) según la reivindicación 9, en el que el sistema (1) comprende además una interfaz de usuario (1211) configurada para recibir un valor umbral de agrupación de un usuario, que responde a mostrar el grafo en una pantalla (33) ; el generador de grafos (126) está configurado además para definir las agrupaciones de nodos mediante la aplicación del valor umbral de agrupación como una distancia máxima dentro de una agrupación, y para mostrar el grafo en la pantalla (33) después de aplicar el valor umbral de agrupación.
11. El sistema (1) según una de las reivindicaciones 9 o 10, en el que el detector de centroide (123) está configurado además para definir la secuencia de ADN asociada con el nodo que tiene la densidad de conectividad más alta en una agrupación, como la secuencia centroide (C) de esa agrupación.
12. El sistema (1) según una de las reivindicaciones 7 a 11, en el que el detector de centroide (123) está configurado además para asignar la anotación de clasificación asociada con una secuencia centroide (C) a secuencias de ADN (111) asociadas con esa secuencia centroide (C) .
13. El sistema (1) según una de las reivindicaciones 7 a 12, en el que el módulo comparador (122) está configurado además para determinar la medida de distancia entre dos secuencias de ADN (111) restando una puntuación ponderada de similitud de una, la puntuación ponderada de similitud se calcula dividiendo la puntuación de similitud entre las dos secuencias de ADN (111) entre la longitud más corta de las dos secuencias de ADN (111) .
14. Un producto de programa informático que comprende medios de código de programa informático para controlar uno o varios procesadores de un sistema informático (1) , de tal manera que el sistema informático (1) realiza el método según una de las reivindicaciones 1 a 6.
El producto de programa informático según la reivindicación 14, que comprende además un medio legible por ordenador que contiene los medios de código de programa informático.
Patentes similares o relacionadas:
Haplotipado y tipado del número de copias mediante el uso de frecuencias alélicas de variantes polimórficas, del 10 de Octubre de 2018, de KATHOLIEKE UNIVERSITEIT LEUVEN: Un método implementado por ordenador para el análisis del material genético de un sujeto, y dicho método comprende: - obtener valores de frecuencias de alelos de variantes […]
Sistema de análisis, método y producto de programa informático para el análisis de muestras biológicas, del 22 de Noviembre de 2017, de F. HOFFMANN-LA ROCHE AG: Sistema de análisis para analizar muestras biológicas, que comprende: - un número N de analizadores (100, 100.1, 100.2, ..., 100.n, ..., 100.N), estando acoplado cada […]
Procedimiento implementado por ordenador y sistema informático para la identificación de organismos, del 19 de Octubre de 2016, de SMARTGENE GMBH: Un procedimiento implementado por ordenador para identificar tipos de organismos a partir de una secuencia genética diana, que comprende: recibir la secuencia […]
PROCEDIMIENTO DE TRAZABILIDAD INDIVIDUAL DE PIEZAS DE IMPLANTOLOGÍA DENTAL Y SISTEMA IDENTIFICATIVO UNITARIO PARA LLEVAR A CABO DICHO PROCEDIMIENTO, del 7 de Julio de 2016, de FERMOINVERS, S.L: Procedimiento de trazabilidad individual de piezas de implantología dental y sistema identificativo unitario para llevar a cabo dicho procedimiento, que comprende: asociar […]
Procedimiento de trazabilidad individual de piezas de implantología dental y sistema identificativo unitario para llevar a cabo dicho procedimiento, del 1 de Julio de 2016, de FERMOINVERS, S.L: Procedimiento de trazabilidad individual de piezas de implantología dental y sistema identificativo unitario para llevar a cabo dicho procedimiento, […]
Métodos para seleccionar medicamentos antidepresivos, del 27 de Abril de 2016, de MAYO FOUNDATION FOR MEDICAL EDUCATION AND RESEARCH: Método para seleccionar un medicamento antidepresivo para un paciente, comprendiendo dicho método: determinar dicho genotipo de un paciente […]
 Métodos para seleccionar medicamentos antidepresivos, del 29 de Enero de 2016, de MAYO FOUNDATION FOR MEDICAL EDUCATION AND RESEARCH: Medio legible por ordenador que contiene instrucciones ejecutables que cuando se ejecutan hacen que un procesador realice operaciones para seleccionar un medicamento […]
Métodos para seleccionar medicamentos antidepresivos, del 29 de Enero de 2016, de MAYO FOUNDATION FOR MEDICAL EDUCATION AND RESEARCH: Medio legible por ordenador que contiene instrucciones ejecutables que cuando se ejecutan hacen que un procesador realice operaciones para seleccionar un medicamento […]
Resolución de fracciones de genoma usando recuento de polimorfismos, del 3 de Diciembre de 2018, de Verinata Health, Inc: Un método para estimar una fracción del ADN fetal en ADN obtenido de un fluido corporal de un individuo embarazado, el método comprendiendo: (a) mapear segmentos de ADN obtenidos […]