Compuestos heterocíclicos condensados, y sus composiciones y usos.
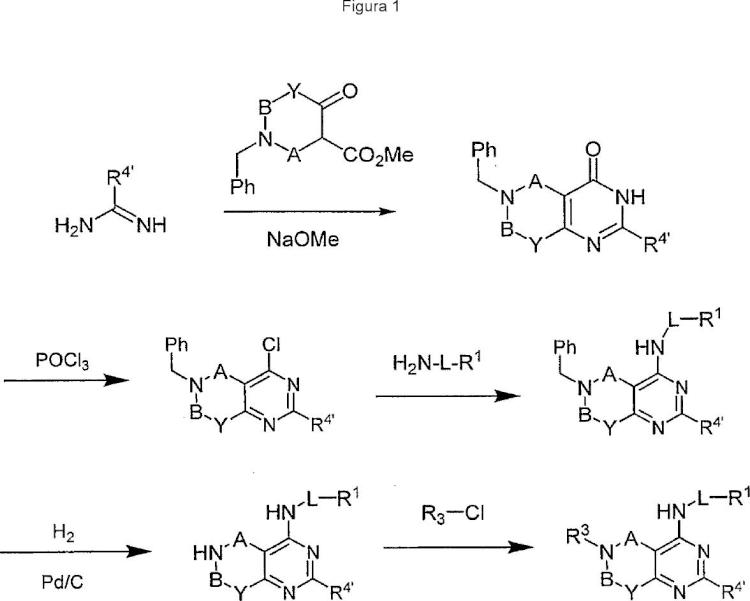
Un compuesto que tiene la fórmula 1:**Fórmula**
en la que
A y B se seleccionan independientemente de CR2'R2',
CO, y CS;
Y es CR2'R2';
W es N;
Z se selecciona de O y NR2;
L es alquileno de C1-C9 o heteroalquileno de C1-C9;
R1 es un grupo carbocíclico o un grupo heterocíclico;
R2 se selecciona de hidrógeno, alquilo de C1-C6, cicloalquilo de C3-C8;
cada R2' se selecciona de hidrógeno, alquilo de C1-C6, cicloalquilo de C3-C8, arilo y aralquilo;
R3 es un grupo carbocíclico o un grupo heterocíclico;
R4' es H; y en la que L, R1, R2, R2' y R3 pueden estar sustituidos o no sustituidos;
o una sal farmacéuticamente aceptable, solvato, estereoisómero, tautómero o variante isotópica del mismo.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/US2006/017614.
Solicitante: Evotec AG.
Inventor/es: KELLY,MICHAEL,G, KINCAID,JOHN, KAUB,CARL J.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A61K31/519 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › condensadas en orto o en peri con heterociclos.
- A61P25/28 A61 […] › A61P ACTIVIDAD TERAPEUTICA ESPECIFICA DE COMPUESTOS QUIMICOS O DE PREPARACIONES MEDICINALES. › A61P 25/00 Medicamentos para el tratamiento de trastornos del sistema nervioso. › de los problemas neurodegenerativos del sistema nervioso central, p. ej. noótropos, activadores del conocimiento, medicamentos para el tratamiento del Alzheimer o de otras formas de demencia.
- C07D471/04 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07D COMPUESTOS HETEROCICLICOS (Compuestos macromoleculares C08). › C07D 471/00 Compuestos heterocíclicos que contienen átomos de nitrógeno como únicos heteroátomos del sistema condensado, teniendo al menos un ciclo de seis miembros con un átomo de nitrógeno, no previstos en los grupos C07D 451/00 - C07D 463/00. › Sistemas condensados en orto.
- C07D487/04 C07D […] › C07D 487/00 Compuestos heterocíclicos que contienen átomos de nitrógeno como únicos heteroátomos del ciclo en el sistema condensado, no previstos por los grupos C07D 451/00 - C07D 477/00. › Sistemas condensados en orto.
- C07F9/141 C07 […] › C07F COMPUESTOS ACICLICOS, CARBOCICLICOS O HETEROCICLICOS QUE CONTIENEN ELEMENTOS DISTINTOS DEL CARBONO, HIDROGENO, HALOGENOS, OXIGENO, NITROGENO, AZUFRE, SELENIO O TELURO (porfirinas que contienen metal C07D 487/22; compuestos macromoleculares C08). › C07F 9/00 Compuestos que contienen elementos de los grupos 5 o 15 del sistema periódico. › Esteres de ácidos fosforosos.
PDF original: ES-2552804_T3.pdf
Patentes similares o relacionadas:
Compuestos de heteroaril carboxamida como inhibidores de RIPK2, del 29 de Julio de 2020, de BOEHRINGER INGELHEIM INTERNATIONAL GMBH: Un compuesto de fórmula (I): **(Ver fórmula)** o sus sales farmacéuticamente aceptables, en la que: X es N y Y es CH; o X es CH y Y es N; […]
Compuestos de alquinilbenceno heterocíclicos, y composiciones médicas y usos de los mismos, del 29 de Julio de 2020, de Guangzhou Healthquest Pharma Co., Ltd: Un compuesto de alquinilbenceno heterocíclico que tiene la fórmula (I) y una sal farmacéuticamente aceptable, o estereoisómero del mismo, **(Ver […]
1,5-Dihidro-4H-pirazolo[3,4-d]pirimidin-4-onas y 1,5-dihidro-4H-pirazolo[4,3-c]piridin-4-onas como inhibidores de la PDE1, del 29 de Julio de 2020, de H. LUNDBECK A/S: Un compuesto de fórmula (I) **(Ver fórmula)** en donde Y es N o CH; R1 se selecciona del grupo que consiste en alquilo C2-C8 lineal o ramificado, cicloalquilo […]
Compuestos y procedimientos de uso, del 29 de Julio de 2020, de Medivation Technologies LLC: Un compuesto de fórmula (Aa-1): **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en la que: A representa H, halógeno, amino, […]
Ureas cíclicas como inhibidores de ROCK, del 22 de Julio de 2020, de BRISTOL-MYERS SQUIBB COMPANY: Un compuesto de acuerdo con la Fórmula (I): **(Ver fórmula)** o un enantiómero, un diastereómero, un estereoisómero, un tautómero, una sal farmacéuticamente aceptable […]
Derivado heteroarilo o sal farmacéuticamente aceptable del mismo, método de preparación del mismo y composición farmacéutica para prevenir o tratar enfermedades asociadas con PI3 quinasas, que contiene el mismo como principio activo, del 22 de Julio de 2020, de KOREA RESEARCH INSTITUTE OF CHEMICAL TECHNOLOGY: Un compuesto representado por la fórmula 1, un isómero óptico del mismo o una sal farmacéuticamente aceptable del mismo: **(Ver fórmula)** en la fórmula […]
Derivado de dihidropiridazin-3,5-diona, del 15 de Julio de 2020, de CHUGAI SEIYAKU KABUSHIKI KAISHA: Un compuesto representado por la fórmula (I) o una sal del mismo, o un solvato del compuesto o la sal: **(Ver fórmula)** en donde R1, R4 y R5 se definen […]
Derivados de 5-[2-(piridin-2-ilamino)-1,3-tiazol-5-il]-2,3-dihidro-1H-isoindol-1-ona y su uso como inhibidores dobles de fosfatidilinositol··3-cinasa delta y gamma, del 15 de Julio de 2020, de ASTRAZENECA AB: Compuesto de formula (I) **(Ver fórmula)** donde X es C(O) o SO2; Y es SO2NHR5 o SO2R6; R1 se selecciona de alquilo C1-4, […]