FORMAS CRISTALINAS DE UN COMPUESTO DE DIMETILFENILO.
Forma de base libre cristalina de éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2- hidroxietilamino]me- til}-2,
5-dimetilfenilcarbamoil)etil]piperidin-4-ílico del ácido bifenil-2- ilcarbámico.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/US2007/010120.
Solicitante: THERAVANCE, INC..
Nacionalidad solicitante: Estados Unidos de América.
Dirección: 901 GATEWAY BOULEVARD SOUTH SAN FRANCISCO, CA 94080 ESTADOS UNIDOS DE AMERICA.
Inventor/es: BOLTON,JENNIFER, CHAO,ROBERT, RAPTA,Miroslav, WILLIAMS,Lisa, WILSON,Richard D.
Fecha de Publicación: .
Fecha Solicitud PCT: 24 de Abril de 2007.
Clasificación PCT:
- A61K31/445 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 31/00 Preparaciones medicinales que contienen ingredientes orgánicos activos. › Piperidinas no condensadas, p. ej. piperocaína.
- A61P11/00 A61 […] › A61P ACTIVIDAD TERAPEUTICA ESPECIFICA DE COMPUESTOS QUIMICOS O DE PREPARACIONES MEDICINALES. › Medicamentos para el tratamiento de trastornos del aparato respiratorio.
- C07D211/46 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07D COMPUESTOS HETEROCICLICOS (Compuestos macromoleculares C08). › C07D 211/00 Compuestos heterocíclicos que contienen ciclos hidrogenados de piridina, no condensados con otros ciclos. › que tienen un átomo de hidrógeno como el segundo sustituyente en posición 4.
Países PCT: Austria, Bélgica, Suiza, Alemania, Dinamarca, España, Francia, Reino Unido, Grecia, Italia, Liechtensein, Luxemburgo, Países Bajos, Suecia, Mónaco, Portugal, Irlanda, Eslovenia, Finlandia, Rumania, Chipre, Lituania, Letonia, Ex República Yugoslava de Macedonia, Albania.
PDF original: ES-2371425_T3.pdf
Fragmento de la descripción:
Formas cristalinas de un compuesto de dimetilfenilo.
Antecedentes dé la invención
La presente invención se refiere a nuevas formas cristalinas de base libre de un compuesto de dimetilfenilo, cuyo compuesto se espera que sea útil como agente terapéutico para el tratamiento de enfermedades pulmonares. Esta invención se refiere también a composiciones farmacéuticas que contienen o están preparados a partir de esta forma cristalina, los procesos y productos intermedios útiles para preparar dichas formas cristalinas, y tales formas cristalinas para su uso en, por ejemplo, el tratamiento de una enfermedad pulmonar.
WO 2004/074246 describe el compuesto del ejemplo 72, que tiene una estructura cercana a la del presente compuesto para usar en el tratamiento de enfermedades pulmonares.
La solicitud Internacional de titularidad compartida PCT nº 2007/009925, solicitada en la misma fecha que la presente, y la solicitud provisional nº 60/794,702, solicitada el 25 de abril de 2006, divulgan nuevos compuestos de dialquilfenilo que son útiles como agentes terapéuticos para tratar enfermedades pulmonares, como la enfermedad pulmonar obstructiva crónica (COPD) y el asma. En particular, el compuesto éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2-hidroxietilamino]metil}-2,5-dimetilfenilcarbamoil)etil]piperidin-4-ílico del ácido bifenil-2-ilcarbámico se describe específicamente en esta solicitud como poseedor de actividad tanto de antagonista muscarínico como de agonista del receptor adrenérgico
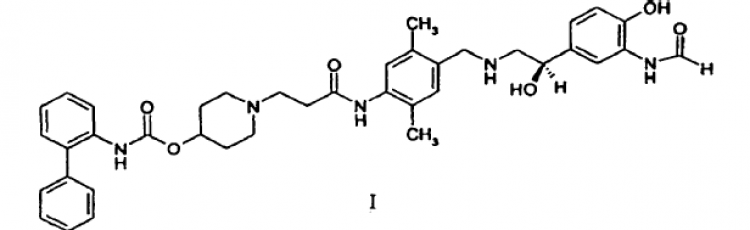
Los agentes terapéuticos útiles para tratar enfermedades pulmonares se administran ventajosamente de forma directa en el tracto respiratorio por inhalación. A este respecto, se han desarrollado varios tipos de dispositivos farmacéuticos de inhalación para administrar agentes terapéuticos por inhalación incluyendo los inhaladores de polvo seco (DPI), inhaladores dosificadores (MDI) e inhaladores nebulizadores. Al preparar composiciones y formulaciones farmacéuticas para el uso en tales dispositivos, es sumamente deseable tener una forma cristalina del agente terapéutico que no sea higroscópica ni delicuescente y que tenga un punto de fusión relativamente alto (es decir, superior a aproximadamente 130ºC) permitiendo así que el material se micronice sin descomposición o pérdida significativas de su cristalinidad.
No se han divulgado formas cristalinas del compuesto de la fórmula I anteriormente. Por consiguiente, existe una necesidad de una forma cristalina estable y no delicuescente del compuesto de la fórmula I que tenga un nivel aceptable de higroscopicidad y un punto de fusión relativamente alto.
Sumario de la invención
La presente invención se refiere a formas cristalinas de base libre de éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2-hidroxietilamino]metil}-2,5-dimetilfenilcarbamoil)etil]piperidin- 4-ílico del ácido bifenil-2-ilcarbámico o un solvato del mismo. Sorprendentemente, se ha descubierto que tales formas cristalinas de base libre del compuesto de la fórmula I no son delicuescentes ni siquiera al ser expuestas a la humedad atmosférica. Además, tales formas cristalinas tienen un nivel aceptable de higroscopicidad y un punto de fusión alto, por ejemplo, superior a aproximadamente 130ºC.
En un aspecto particular, esta invención se refiere a una forma de base libre cristalina de éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2-hidroxietilamino]metil}-2,5-dimetilfenil-carbamoil)etil]piperidin-4-ílico del ácido bifenil-2-ilcarbámico o un solvato del mismo en forma micronizada.
Entre otros usos, una forma de base libre cristalina del compuesto de la fórmula I sirve para preparar composiciones farmacéuticas que se esperan que sean útiles para tratar enfermedades pulmonares. Por consiguiente, en otro de los aspectos de su composición, la presente invención se refiere a una composición farmacéutica que comprende un soporte farmacéuticamente aceptable y una forma de base libre cristalina de éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2-hidroxietilamino]metil}-2,5-dimetilfenilcarbamoil)etil]piperidin-4-ílico del ácido bifenil-2-ilcarbámico o un solvato del mismo.
Si se desea, las formas cristalinas de la presente invención pueden administrarse en combinación con otros agentes terapéuticos, como un agente antiinflamatorio esteroideo.
En consecuencia, en otro de los aspectos de su composición, esta invención se refiere a una composición farmacéutica que comprende (a) una forma de base libre cristalina de éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2-hidroxietilamino]metil}-2,5-dimetilfenilcarbamoil)-etil]piperidin-4-ílico del ácido bifenil-2-ilcarbámico o un solvato del mismo; y (b) un segundo agente terapéutico. En aún otro de los aspectos de su composición, esta invención se refiere a una composición farmacéutica que comprende (a) una forma de base libre cristalina de éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2-hidroxietilamino]metil}-2,5-dimetilfenilcarbamoil)-etil]piperidin-4-ílico del ácido bifenil-2-ilcarbámico o un solvato del mismo; y (c) un soporte farmacéuticamente aceptable.
En aún en otro de los aspectos de su composición, esta invención se refiere a una combinación de agentes terapéuticos, la combinación comprendiendo (a) una forma de base libre cristalina de éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2-hidroxietilamino]metil}-2,5-dimetilfenilcarbamoil)etil]piperidin-4-ílico del ácido bifenil-2-ilcarbámico o un solvato del mismo; y (b) un segundo agente terapéutico. En otro de los aspectos de su composición, esta invención se refiere a una combinación de composiciones farmacéuticas, la combinación comprendiendo (a) una composición farmacéutica que comprende una forma de base libre cristalina de éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2-hidroxietilamino]metil}-2,5-dimetilfenilcarbamoil)etil]piperidin-4-ílico del ácido bifenil-2-ilcarbámico o un solvato del mismo y un soporte farmacéuticamente aceptable; y (b) una composición farmacéutica que comprende un segundo agente terapéutico y un soporte farmacéuticamente aceptable.
El compuesto de la fórmula I posee tanto actividad de agonista del receptor adrenérgico
Por consiguiente, en uno de sus aspectos, esta invención se refiere a una forma de base libre cristalina de un compuesto de la fórmula I para usar en un método para tratar una enfermedad pulmonar, el método comprendiendo administrar a un paciente necesitado de tratamiento una cantidad terapéuticamente efectiva de una forma de base libre cristalina de éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2-hidroxietilamino]metil}-2,5-dimetilfenil-carbamoil)etil]piperidin-4-ílico del ácido bifenil-2-ilcarbámico o un solvato del mismo.
Esta invención también se refiere a una forma de base libre cristalina de un compuesto de la fórmula I para su uso en un método para tratar la enfermedad pulmonar obstructiva crónica o el asma, el método comprendiendo administrar a un paciente una cantidad terapéuticamente efectiva de una forma de base libre cristalina de éster 1-[2-(4-{[(R)-2-(3- formilamino-4-hidroxifenil)-2-hidroxietilamino]metil}-2,5-dimetilfenilcarbamoil)etil]piperidin-4- ílico del ácido bifenil-2-ilcarbámico o un solvato del mismo.
Adicionalmente, en otro de los aspectos de sus métodos, esta invención se refiere a una forma de base libre cristalina de un compuesto de la fórmula I para usar en un método para producir broncodilatación en un mamífero, el método comprendiendo administrar a un mamífero una cantidad que produzca la broncodilatación de una forma de base libre cristalina de éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2-hidroxietilamino]metil}-2,5-dimetilfenil-carbamoil)etil]piperidin-4-ílico... [Seguir leyendo]
Reivindicaciones:
1. Forma de base libre cristalina de éster 1-[2-(4-{[(R)-2-(3-formilamino-4-hidroxifenil)-2- hidroxietilamino]me- til}-2,5-dimetilfenilcarbamoil)etil]piperidin-4-ílico del ácido bifenil-2- ilcarbámico.
2. Forma de base libre cristalina de la reivindicación 1 que tiene la Forma I, que está caracterizada por un patrón de difracción de rayos x en polvo que tiene picos de difracción en valores 2θ de 17,7
3. Forma de base libre cristalina de la reivindicación 1 que tiene la Forma II, que está caracterizada por un patrón de difracción de rayos x en polvo que tiene picos de difracción en valores 2θ de 20,70
4. Forma de base libre cristalina de la reivindicación 1 que tiene la Forma III, que está caracterizada por un patrón de difracción de rayos x en polvo que tiene picos de difracción en valores 2θ de 15,50
5. Forma de base libre cristalina de la reivindicación 1 en forma micronizada.
6. Composición farmacéutica que comprende un soporte farmacéuticamente aceptable y una forma de base libre cristalina de cualquiera de las reivindicaciones 1 a 5.
7. Composición farmacéutica que comprende:
8. Solución salina isotónica preparada disolviendo la forma de base libre cristalina de cualquiera de las reivindicaciones 1 a 5 en solución salina acuosa isotónica; en donde la solución tiene un pH en el rango de 4 a 6.
9. Combinación de agentes terapéuticos que comprende:
10. Proceso para preparar la forma de base libre cristalina de la reivindicación 1, el proceso comprendiendo:
11. Proceso para preparar la forma de base libre cristalina de la reivindicación 1, el proceso comprendiendo:
12. Forma de base libre cristalina según cualquiera de las reivindicaciones 1 a 5 para su uso en terapia.
13. Uso de una forma de base libre cristalina de cualquiera de las reivindicaciones 1 a 5 para la fabricación de un medicamento para tratar una enfermedad pulmonar.
Patentes similares o relacionadas:
Compuestos y procedimientos de uso, del 29 de Julio de 2020, de Medivation Technologies LLC: Un compuesto de fórmula (Aa-1): **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en la que: A representa H, halógeno, amino, […]
Triple combinación de antagonistas del receptor 5-HT6 puros, inhibidores de la acetilcolinesterasa y antagonista del receptor NMDA, del 15 de Julio de 2020, de SUVEN LIFE SCIENCES LIMITED: Una combinación que comprende un antagonista del receptor 5-HT6 puro, un inhibidor de la acetilcolinesterasa y un antagonista del receptor NMDA, en el que el antagonista […]
Combinación de agonistas inversos del receptor de histamina-3 con inhibidores de acetilcolinesterasa, del 24 de Junio de 2020, de SUVEN LIFE SCIENCES LIMITED: Una combinación que comprende un agonista inverso del receptor de histamina-3 y un inhibidor de acetilcolinesterasa; en donde el agonista inverso del receptor de histamina-3 […]
Compuestos inhibidores de la acetilcolinesterasa y agonistas de los receptores serotoninérgicos 5HT4, con efecto paramnesiante, sus procedimientos de preparación y composiciones farmacéuticas que los contienen, del 10 de Junio de 2020, de Université de Caen: Compuesto de fórmula general (I): **(Ver fórmula)** en la que: X representa un átomo de hidrógeno, o un átomo de halógeno […]
Nuevos derivados de piperazina y piperidina, síntesis y uso de los mismos en la inhibición de la oligomerización de VDAC, la apoptosis y la disfunción mitocondrial, del 3 de Junio de 2020, de The National Institute for Biotechnology in the Negev Ltd: Compuesto de Fórmula general (Id): **(Ver fórmula)** en la que L2 es un grupo de enlace seleccionado del grupo que consiste en un alquilamidileno […]
Compuestos de benzaldehído sustituidos y métodos para su uso en el aumento de la oxigenación tisular, del 27 de Mayo de 2020, de Global Blood Therapeutics, Inc: Un compuesto de Fórmula (I): **(Ver fórmula)** o un tautómero o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento […]
Composición farmacéutica en comprimido que comprende bilastina, del 27 de Mayo de 2020, de Alfred E. Tiefenbacher (GmbH & Co. KG): Una composición farmacéutica en forma de comprimido, que comprende a) una forma cristalina de bilastina, en donde la forma cristalina tiene picos característicos a 6,53, […]
Nuevas composiciones para prevenir y/o tratar trastornos degenerativos del sistema nervioso central, del 27 de Mayo de 2020, de AMICUS THERAPEUTICS, INC: Un compuesto de Fórmula III: **(Ver fórmula)** en donde: R1 es C(R2)(R3)(R4); R2 es hidrógeno, -OH o halógeno; R3 es hidrógeno, -OH, halógeno o -CH3; […]