Factor de coagulación VIIa modificado con semivida prolongada.
Un polipéptido fusionado con albúmina que comprende al menos un polipéptido de Factor VII o Factor VIIa fusionado con albúmina,
en el que al menos uno de estos polipéptidos de Factor VII o Factor VIIa está localizado en el extremo N-terminal de la proteína de fusión y en el que la proteína de fusión tiene actividad biológica de Factor VII/VIIa y en el que un enlazador peptídico separa el resto del Factor VII o del Factor VIIa en el extremo N-terminal de la proteína de fusión, del resto de albúmina, comprendiendo dicho enlazador al menos 25 aminoácidos y unidades de repetición que comprenden glicina y serina.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/EP2007/000937.
Solicitante: CSL BEHRING GMBH.
Nacionalidad solicitante: Alemania.
Dirección: EMIL-VON-BEHRING-STRASSE 76 35041 MARBURG ALEMANIA.
Inventor/es: WEIMER, THOMAS, DR., LANG, WIEGAND, WORMSBACHER, WILFRIED, SCHULTE,STEFAN,DR, LIEBING,UWE, KRONTHALER,ULRICH.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A61K38/48 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 38/00 Preparaciones medicinales que contienen péptidos (péptidos que contienen ciclos beta-lactama A61K 31/00; dipéptidos cíclicos que no tienen en su molécula ningún otro enlace peptídico más que los que forman su ciclo, p. ej. piperazina 2,5-dionas, A61K 31/00; péptidos basados en la ergolina A61K 31/48; que contienen compuestos macromoleculares que tienen unidades aminoácido repartidas estadísticamente A61K 31/74; preparaciones medicinales que contienen antígenos o anticuerpos A61K 39/00; preparaciones medicinales caracterizadas por los ingredientes no activos, p. ej. péptidos como soportes de fármacos, A61K 47/00). › que actúan sobre enlaces peptídicos (3.4).
- C07K14/76 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07K PEPTIDOS (péptidos que contienen β -anillos lactamas C07D; ipéptidos cíclicos que no tienen en su molécula ningún otro enlace peptídico más que los que forman su ciclo, p. ej. piperazina diones-2,5, C07D; alcaloides del cornezuelo del centeno de tipo péptido cíclico C07D 519/02; proteínas monocelulares, enzimas C12N; procedimientos de obtención de péptidos por ingeniería genética C12N 15/00). › C07K 14/00 Péptidos con más de 20 aminoácidos; Gastrinas; Somatostatinas; Melanotropinas; Sus derivados. › Albúminas.
- C07K19/00 C07K […] › Péptidos híbridos (Inmoglobulinas híbridas compuestas solamente de inmoglobulinas C07K 16/46).
- C12N15/62 C […] › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12N MICROORGANISMOS O ENZIMAS; COMPOSICIONES QUE LOS CONTIENEN; PROPAGACION, CULTIVO O CONSERVACION DE MICROORGANISMOS; TECNICAS DE MUTACION O DE INGENIERIA GENETICA; MEDIOS DE CULTIVO (medios para ensayos microbiológicos C12Q 1/00). › C12N 15/00 Técnicas de mutación o de ingeniería genética; ADN o ARN relacionado con la ingeniería genética, vectores, p. ej. plásmidos, o su aislamiento, su preparación o su purificación; Utilización de huéspedes para ello (mutantes o microorganismos modificados por ingeniería genética C12N 1/00, C12N 5/00, C12N 7/00; nuevas plantas en sí A01H; reproducción de plantas por técnicas de cultivo de tejidos A01H 4/00; nuevas razas animales en sí A01K 67/00; utilización de preparaciones medicinales que contienen material genético que es introducido en células del cuerpo humano para tratar enfermedades genéticas, terapia génica A61K 48/00; péptidos en general C07K). › Secuencias de ADN que codifican proteínas de fusión.
- C12N9/64 C12N […] › C12N 9/00 Enzimas, p. ej. ligasas (6.); Proenzimas; Composiciones que las contienen (preparaciones para la limpieza de los dientes que contienen enzimas A61K 8/66, A61Q 11/00; preparaciones de uso médico que contienen enzimas A61K 38/43; composiciones detergentes que contienen enzimas C11D ); Procesos para preparar, activar, inhibir, separar o purificar enzimas. › que provienen de tejido animal, p. ej. renina.
PDF original: ES-2504517_T3.pdf




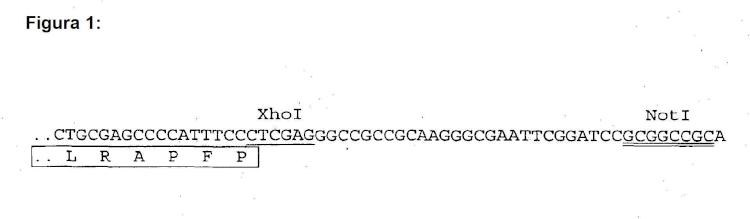
Fragmento de la descripción:
Factor de coagulación VIIa modificado con semivida prolongada Campo de la invención:
La presente invención se refiere al campo de polipéptidos de Factor VII (FVII) y Factor VIIa (FVIIa) enlazados con albúmina. Más específicamente, la invención se refiere a secuencias de ADNc que codifican el Factor VII y el Factor VIIa humanos y derivados, fusionados genéticamente con un ADNc que codifica seroalbúmina humana, en donde al menos uno de tales polipéptidos de Factor VII o Factor VIIa está localizado en el extremo N-terminal de la proteína de fusión y en donde un enlazador peptídico separa el resto del Factor VII o del Factor VIIa en el extremo N-terminal de la proteína de fusión del resto de albúmina, comprendiendo dicho enlazador al menos 25 aminoácidos y unidades de repetición que comprenden glicina y serina, y a procedimientos para la preparación de tales proteínas recombinantes y sus derivados. La invención también incluye un vector de transferencia para uso en terapia génica humana que comprende tales secuencias de ADN modificadas.
Antecedentes de la invención:
Factor VII y Factor VIIa La hemofilia A es un trastorno hemorrágico hereditario. Es el resultado de una carencia del Factor VIII de la coagulación sanguínea, ligada al cromosoma X y afecta casi exclusivamente a varones con una incidencia de entre uno y dos individuos por cada 10.000. El defecto del cromosoma X es transmitido por mujeres portadoras no siendo ellas mismas hemofílicas. La manifestación clínica de la hemofilia A es un aumento de la tendencia a la hemorragia. Antes de la introducción del tratamiento con concentrados de Factor VIII, la esperanza media de vida de una persona con hemofilia grave era menos de 20 años. El uso de concentrados de Factor VIII procedentes del plasma y posteriormente de formas recombinantes del Factor VIII, ha mejorado considerablemente la situación de los pacientes con hemofilia, incrementando en gran medida la esperanza media de vida, proporcionando a la mayoría de ellos la posibilidad de tener una vida más o menos normal. La hemofilia B que es 5 veces menos prevalente que la hemofilia A está causada por un Factor IX no funcional o ausente y se trata con concentrados de Factor IX procedentes del plasma o una forma recombinante de Factor IX. Tanto en la hemofilia A como en la hemofilia B, el problema médico más grave en el tratamiento de la enfermedad es la generación de aloanticuerpos contra los factores de sustitución. Hasta un 30% de todos los pacientes con hemofilia A desarrollan anticuerpos contra el Factor VIII. Los anticuerpos contra el Factor IX se producen en menor medida pero con consecuencias más graves, ya que son menos susceptibles a una terapia de inducción de tolerancia inmune.
El modelo actual de coagulación establece que el agente fisiológico desencadenante de la coagulación es la formación de un complejo entre el Factor tisular (FT) y el Factor VIIa (FVIIa) en la superficie de células que expresan FT, que se encuentran normalmente fuera del sistema vascular. Esto conduce a la activación del Factor IX y el Factor X que generan a la larga cierta cantidad de trombina. En un bucle de retroalimentación positiva, la trombina activa el Factor VIII y el Factor IX, la denominada vía "intrínseca" de la cascada de coagulación de la sangre, amplificando de este modo la generación de Factor Xa, que es necesario para la generación de una ráfaga de trombina completa para lograr la hemostasia completa. Se mostró que mediante la administración de concentraciones suprafisiológicas de Factor VIIa, se consigue una hemostasia evitando la necesidad de Factor VIIIa y Factor IXa. La clonación del ADNc del Factor VII (documento US 4.784.950) hizo posible el desarrollo de Factor VII activado como agente farmacéutico. El Factor VIIa se administró con éxito por primera vez en 1988. Desde entonces el número de indicaciones de Factor VIIa ha crecido de manera constante mostrando que tiene potencial para convertirse en un agente hemostático universal para detener el sangrado (Erhardtsen, 2002) . Sin embargo, la corta semivida del Factor VIIa de aproximadamente 2 horas, está limitando su aplicación.
FVII es una glicoproteína de cadena única con un peso molecular de aproximadamente 50 kDa, que es secretada por las células hepáticas en el torrente sanguíneo como un zimógeno inactivo de 406 aminoácidos. Contiene 10 residuos de ácido γ-carboxi-glutámico (posiciones 6, 7, 14, 16, 19, 20, 25, 26, 29 y 35) localizados en el dominio Gla N-terminal de la proteína. Los residuos Gla requieren vitamina K para su biosíntesis. Dos dominios de factor de crecimiento epidérmico seguidos por un dominio de proteasa de serina de tipo tripsina, se encuentran en posición Cterminal con respecto al dominio Gla. Otras modificaciones postraduccionales de FVII incluyen la hidroxilación (Asp 63) , glicosilación de tipo N (Asn145 y Asn322) así como de tipo O (Ser52 y Ser60) .
FVII se convierte en su forma activa Factor VIIa mediante proteolisis del enlace peptídico único en Arg152-Ile153 que conduce a la formación de dos cadenas polipeptídicas, una cadena ligera N-terminal (24 kDa) y una cadena pesada C-terminal (28 kDa) , que se mantienen unidas por un puente disulfuro. En contraste con otros factores de coagulación dependientes de la vitamina K, no se ha descrito para FVII un péptido de activación, que se escinde durante la activación de estos otros factores de coagulación dependientes de la vitamina K. El sitio de escisión Arg152-Ile153 y algunos aminoácidos aguas abajo muestran homología con el sitio de escisión para la activación de otros polipéptidos dependientes de vitamina K.
Para la consecución de la conformación activa del Factor VIIa, es esencial la formación de un puente salino después de la escisión para activación entre Ile153 y Asp343. La escisión para activación del Factor VII se puede conseguir in
vitro con el Factor Xa, Factor XIIa, Factor IXa, Factor VIIa, proteasa activadora del factor siete (FSAP) y trombina. Mollerup et al. (Biotechnol. Bioeng. (1995) 48: 501-505) informaron que también tiene lugar alguna escisión en la cadena pesada en Arg290 y/o Arg315.
El Factor VII está presente en el plasma en una concentración de 500 ng/ml. Un 1%, por ejemplo 5 ng/ml de Factor VII está presente como Factor VIIa. La semivida plasmática del Factor VII se encontró que era de aproximadamente 4 horas y la del Factor VIIa de aproximadamente 2 horas. Aunque la semivida del Factor VIIa de 2 horas es comparativamente larga para un factor de coagulación activado (que para otros factores de coagulación activados es más del orden de minutos, debido a una inhibición irreversible con serpinas como antitrombina III) , sin embargo, esto constituye un grave inconveniente para el uso terapéutico del Factor VIIa, puesto que conlleva la necesidad de múltiples inyecciones i.v. o una infusión continua para conseguir la hemostasia. Esto da lugar a un tratamiento muy costoso e incomodidad para el paciente. Hasta ahora no está disponible comercialmente ninguna preparación farmacéutica de un Factor VIIa con una semivida plasmática mejorada, ni se ha publicado ningún dato que muestre variantes de FVII/FVIIa con una semivida prolongada in vivo. Como el Factor VII/VIIa tiene el potencial de poder ser utilizado como un agente hemostático universal, todavía existe una gran necesidad médica de desarrollar formas del Factor VIIa que tengan una semivida funcional in vivo más larga.
Ballance et al. (documento WO 01/79271) describe polipéptidos de fusión de múltiples proteínas terapéuticas diferentes o variantes y/o fragmentos de dichas proteínas terapéuticas que, cuando se fusionan con seroalbúmina humana o variantes y/o fragmentos de dicha albúmina, tendrán probablemente una semivida funcional incrementada in vivo y una vida útil prolongada. Se describen largas listas de potenciales parejas de fusión sin mostrar con datos experimentales para casi la totalidad de estas proteínas que los respectivos polipéptidos fusionados con albúmina, realmente conservan la actividad biológica de la pareja de fusión proteica terapéutica y tienen propiedades mejoradas. Según el documento WO 01/79271, además, cada miembro de la lista de proteínas terapéuticas se puede fusionar con orientaciones muy diferentes con la albúmina, por ejemplo, dos moléculas de la proteína terapéutica fusionada una con el extremo N-terminal y la otra con el extremo C-terminal de la albúmina, o una molécula de la proteína terapéutica fusionada con el extremo N-terminal o C-terminal de la albúmina, o también múltiples regiones de cada proteína fusionadas con múltiples regiones de la otra. Entre las múltiples proteínas terapéuticas enumeradas en el documento WO 01/79271 como potenciales... [Seguir leyendo]
Reivindicaciones:
1. Un polipéptido fusionado con albúmina que comprende al menos un polipéptido de Factor VII o Factor VIIa fusionado con albúmina, en el que al menos uno de estos polipéptidos de Factor VII o Factor VIIa está localizado en el extremo N-terminal de la proteína de fusión y en el que la proteína de fusión tiene actividad biológica de Factor VII/VIIa y en el que un enlazador peptídico separa el resto del Factor VII o del Factor VIIa en el extremo N-terminal de la proteína de fusión, del resto de albúmina, comprendiendo dicho enlazador al menos 25 aminoácidos y unidades de repetición que comprenden glicina y serina.
2. Un polipéptido fusionado con albúmina según la reivindicación 1, en el que la proteína de fusión tiene al menos 25% de actividad biológica específica y molar de Factor VII/VIIa en comparación con el Factor VII o el Factor VIIa de tipo silvestre no fusionado respectivo.
3. Un polipéptido fusionado con albúmina que comprende un polipéptido de Factor VII o Factor VIIa según la reivindicación 1 o 2, en el que la proteína de fusión tiene una semivida plasmática funcional incrementada in vivo en comparación con un Factor VII o un Factor VIIa no fusionado.
4. Un polipéptido fusionado con albúmina que comprende un polipéptido de Factor VII o Factor VIIa según la reivindicación 3, en el que la proteína de fusión tiene una semivida funcional in vivo que se incrementa al menos 100% en comparación con un Factor VII o un Factor VIIa no fusionado.
5. El polipéptido fusionado con albúmina según las reivindicaciones 1 a 4, en el que el enlazador contiene un sitio de escisión para proteasa.
6. El polipéptido fusionado con albúmina según la reivindicación 5, en el que el sitio de escisión se puede escindir con una proteasa de la coagulación seleccionada a partir del grupo que consiste en Factor IIa, Factor IXa, Factor Xa, Factor XIa, Factor XIIa, proteína C activada, elastasa o calicreína.
7. El polipéptido fusionado con albúmina según las reivindicaciones 1 a 6, en el que el enlazador se modifica mediante la inserción de sitios para modificaciones postraduccionales y en el que la modificación postraduccional comprende uno o varios sitios de N-glicosilación de la estructura Asn -X -Ser/Thr, en donde X designa cualquier aminoácido excepto prolina.
8. El polipéptido fusionado con albúmina según la reivindicación 1 a 7, en el que dicho polipéptido fusionado con albúmina se modifica de modo que la modificación comprende añadir mediante inserción al menos parte del péptido de activación de un polipéptido diferente dependiente de vitamina K, o añadir mediante inserción un análogo de dicho péptido de activación del polipéptido diferente dependiente de vitamina K.
9. Los polipéptidos fusionados con albúmina según las reivindicaciones 1 a 8, en los que el resto del polipéptido del Factor VII o del Factor VIIa tiene actividad procoagulante.
10. El polipéptido fusionado con albúmina según las reivindicaciones 1 a 9, para uso como medicamento.
11. Una composición farmacéutica que comprende una cantidad eficaz del polipéptido fusionado con albúmina según una cualquiera de las reivindicaciones 1 a 9, junto con un vehículo o un excipiente farmacéuticamente aceptable.
12. Una composición farmacéutica según la reivindicación 11, para uso en el tratamiento o la prevención de trastornos hemorrágicos, en donde el trastorno hemorrágico es preferentemente hemofilia A.
13. Una composición farmacéutica según la reivindicación 12, en la que el polipéptido fusionado con albúmina se administra por vía intravenosa, subcutánea, intramuscular, intraperitoneal, intracerebral, intrapulmonar, intranasal
o transdérmica.
14. Una molécula de ácido nucleico en donde dicha molécula de ácido nucleico comprende una secuencia de polinucleótidos que codifica el polipéptido fusionado con albúmina según las reivindicaciones 1 a 9, en donde dicha secuencia de polinucleótidos se encuentra en el extremo 3' de una secuencia de nucleótidos que codifica un propéptido que media en la carboxilación gamma de la parte fusionada del Factor VII/VIIa.
15. Un plásmido o un vector en donde dicho plásmido o vector comprende la molécula de ácido nucleico según la reivindicación 14.
16. Un plásmido o un vector según la reivindicación 15, en donde dicho plásmido o vector es i) un vector de expresión o ii) un vector de transferencia para uso en terapia génica.
17. Una célula hospedadora en donde dicha célula hospedadora comprende la molécula de ácido nucleico según la reivindicación 14 a 16.
18. Un método para preparar un polipéptido fusionado con albúmina según la reivindicación 1 a 9, en donde di
cho método comprende:
a. proporcionar un ácido nucleico que comprende una secuencia de nucleótidos que codifica el polipéptido fusionado con albúmina que se puede expresar en un organismo;
b. expresar el ácido nucleico en el organismo para formar un polipéptido fusionado con albúmina; y purificar el polipéptido fusionado con albúmina.
Patentes similares o relacionadas:
Cadena ligera de enteroquinasa modificada, del 22 de Julio de 2020, de NOVO NORDISK A/S: Un análogo de la cadena ligera de la enteroquinasa bovina que comprende una secuencia de aminoácidos establecida en la SEQ ID NO: 1, en donde dicho análogo comprende […]
Detección de interacciones proteína a proteína, del 15 de Julio de 2020, de THE GOVERNING COUNCIL OF THE UNIVERSITY OF TORONTO: Un método para medir cuantitativamente la fuerza y la afinidad de una interacción entre una primera proteína de membrana o parte de la misma y una […]
Métodos y composiciones para ingeniería genómica, del 3 de Junio de 2020, de Sangamo Therapeutics, Inc: Una pareja de nucleasas de dedo de zinc (ZFN) que comprende una ZFN izquierda y una ZFN derecha, comprendiendo cada ZFN un dominio de escisión […]
Métodos y composiciones para escisión dirigida y recombinación, del 20 de Mayo de 2020, de Sangamo Therapeutics, Inc: Un método in vitro para la escisión selectiva de un gen HLA clase I, un gen HLA que codifica una proteína de clase 1 del Complejo de Histocompatibilidad Mayor (MHC) […]
Antígenos de coagulasa estafilocócica y métodos para su uso, del 13 de Mayo de 2020, de UNIVERSITY OF CHICAGO: Una composición inmunógena que comprende al menos dos dominios 1-2 de coagulasa estafilocócica diferentes, en donde cada uno de los al menos dos dominios […]
Reconocimiento de unión a diana celular mediante un agente bioactivo usando transferencia de energía de resonancia de bioluminiscencia intracelular, del 6 de Mayo de 2020, de PROMEGA CORPORATION: Un sistema de ensayo que comprende: (a) una biblioteca de agentes bioactivos, cada uno de los cuales está fijado a un fluoróforo; (b) una diana celular fusionada a […]
Utilización diagnóstica de un polipéptido de fusión que comprende una proteína vírica y un enzima MGMT, del 15 de Abril de 2020, de INSTITUT PASTEUR: Utilización in vitro de un polipéptido de fusión que comprende una proteína vírica y i) el enzima 6-metilguanina-ADN-metiltransferasa (MGMT, EC 2.1.1.63) o un homólogo […]
Etiqueta de epítopo y método de detección, captura y/o purificación de polipéptidos etiquetados, del 15 de Abril de 2020, de ChromoTek GmbH: Péptido epítopo aislado que tiene de 12 a 25 aminoácidos, en donde la secuencia de aminoácidos comprende una secuencia según se define en SEQ ID NO: 32 (X1X2RX4X5AX7SX9WX11X12), […]