COMPRIMIDO DE LIBERACION MODIFICADA DE HIDROCLORURO DE BUPROPION.
Comprimido de liberación modificada que comprende:
(i) un núcleo que comprende una cantidad eficaz de una sal farmacéuticamente aceptable de bupropión,
y excipientes convencionales;
(ii) un primer recubrimiento de liberación controlada que rodea dicho núcleo en el que dicho recubrimiento de liberación controlada comprende un polímero formador de película permeable al agua insoluble en agua, un plastificante y un polímero soluble en agua y en el que la relación del polímero formador de película permeable al agua insoluble en agua:plastificante:polímero soluble en agua es de 3:1:4 a 5:1:3; y
(iii) una barrera de humedad que rodea dicho primer recubrimiento de liberación controlada, en el que la barrera de humedad comprende un polímero entérico, un plastificante y un mejorador de la permeación, en el que el mejorador de la permeación está presente en una cantidad de 20% a 40% en peso del peso seco de la barrera de humedad;
en el que el comprimido de liberación modificada es bioequivalente y exhibe un perfil de disolución tal que después de aproximadamente 2 horas se libera no más de aproximadamente 20% del contenido de bupropión, después de aproximadamente 4 horas se libera 15% a 45% del contenido de bupropión, después de aproximadamente 8 horas se libera 40% a 90% del contenido de bupropión y después de aproximadamente 16 horas se libera no menos de aproximadamente 80% del contenido de bupropión
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/US2003/024700.
Solicitante: BIOVAIL LABORATORIES INTERNATIONAL SRL.
Nacionalidad solicitante: Barbados.
Dirección: WELCHES,CHRIST CHURCH 17154.
Inventor/es: MAES, PAUL, OBEREGGER,WERNER, ERADIRI,OKPO, ZHOU,FANG.
Fecha de Publicación: .
Fecha Concesión Europea: 6 de Enero de 2010.
Clasificación Internacional de Patentes:
- A61K9/28K
Clasificación PCT:
- A61K9/28 NECESIDADES CORRIENTES DE LA VIDA. › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › A61K 9/00 Preparaciones medicinales caracterizadas por un aspecto particular. › Grageas; Píldoras o comprimidos con revestimientos.
Clasificación antigua:
Fragmento de la descripción:
Comprimido de liberación modificada de hidrocloruro de bupropión.
Campo de la invención
La presente invención se refiere a un comprimido de liberación modificada de sales farmacéuticamente aceptables de bupropión, preferentemente hidrocloruro de bupropión.
Antecedentes
El bupropión es un antidepresivo no relacionado químicamente con inhibidores tricíclicos, tetracíclicos, selectivos de reabsorción de serotonina (SSRI), u otros agentes antidepresivos conocidos. El fármaco se asemeja a un psicoestimulante en términos de sus perfiles neuroquímico y conductual in vivo, pero no produce fiablemente efectos afines a los estimulantes en los humanos a las dosis prescritas clínicamente. Su estructura se asemeja estrechamente a la del dietilpropión y está relacionada con las feniletilaminas. El mismo se designa como (
El mecanismo neuroquímico del efecto antidepresivo del bupropión no se conoce bien. El bupropión no inhibe la monoamina-oxidasa. El bupropión afecta a los productos químicos en el interior del cerebro que utilizan los nervios para enviar mensajes unos a otros. Estos mensajeros químicos se denominan neurotransmisores. Los neurotransmisores que son liberados por los nervios son recogidos de nuevo por los nervios que los liberan para su reutilización (se hace referencia a esto como reabsorción). Muchos expertos creen que la depresión está causada por un desequilibrio entre las cantidades de neurotransmisores que se liberan. Se cree que el bupropión trabaja inhibiendo la reabsorción de los neurotransmisores dopamina, serotonina, y norepinefrina, una acción que da como resultado la producción de más dopamina, serotonina, y norepinefrina disponibles para transmitir mensajes a otros nervios. De acuerdo con ello, el bupropión es único en el sentido de que su efecto principal tiene lugar sobre la dopamina, un efecto que no es compartido por los SSRI (por ejemplo paroxetina (Paxil®), fluoxetina (Prozac®), sertralina (Zoloft®)) o los antidepresivos tricíclicos o TCA (por ejemplo amitriptilina (Elavil®), imipramina (Tofranil®), desipramina (Norpramin®)).
El Wellbutrin® y el Wellbutrin® SR se utilizan para el tratamiento de la depresión. El Zyban® ha sido aprobado como ayuda para aquellos pacientes que quieren dejar de fumar. El Wellbutrin®, la formulación de liberación inmediata de bupropión, se dosifica 3 veces al día, preferentemente con 6 o más horas entre las dosis. Para los pacientes que requieren más de 300 mg de bupropión al día, cada dosis no debería exceder de 150 mg. Esto requiere la administración de los comprimidos por lo menos cuatro veces al día con por lo menos 4 horas entre las dosis. La formulación de liberación inmediata da como resultado más de un 75% de liberación del bupropión en el medio de disolución en aproximadamente 45 minutos, y uno de los efectos secundarios principales de bupropión ha sido la incidencia de ataques, que parece en parte estar fuertemente asociada con la liberación inmediata del bupropión en el sistema. De acuerdo con ello, se desarrollaron productos de liberación sostenida para evitar la incidencia de ataques. Los productos de liberación sostenida se dosifican dos veces al día.
En general, el cumplimiento del paciente es un problema con las medicaciones que requieren un régimen de dosificación múltiple y es especialmente problemática con los individuos deprimidos. Si bien las formulaciones de liberación sostenida han simplificado el régimen de dosificación y aumentado el cumplimiento de los pacientes, existe todavía espacio para la simplificación adicional del régimen de dosificación y mejorar ulteriormente la adhesión de los pacientes al régimen de dosificación. El desarrollo de una formulación de bupropión con liberación modificada aprobada y estable una vez al día sería un avance en la técnica.
Las formas de comprimido de liberación sostenida de bupropión han sido descritas en la técnica anterior. La patente U.S. nº 4687660 describe un comprimido formado por un núcleo y un recubrimiento, en el que el núcleo comprende hidrocloruro de bupropión junto con uno o más excipientes y opcionalmente un agente mejorador de la ósmosis y en el que el recubrimiento comprende un polímero formador de película, permeable al agua e insoluble en agua (tal como acetato de celulosa), un agente formador de poros (tal como lactosa y carbonato de sodio impalpable), y opcionalmente un denominado agente mejorador de la permeabilidad al agua (tal como polietilenglicol) y de nuevo opcionalmente un plastificante.
Las patentes U.S. nº. 5.358.970 y 5.427.798 describen una formulación de liberación sostenida de hidrocloruro de bupropión basada en tecnología de matrices. El término matriz se refiere a un comprimido en la cual el fármaco está embebido en un excipiente que produce un núcleo no disgregante denominado una matriz. La difusión del fármaco ocurre a través de este núcleo. Dado que el hidrocloruro de bupropión es inestable, el producto descrito en las dos patentes anteriores requiere un estabilizador para conseguir una estabilidad suficiente. Este estabilizador es un compuesto ácido, preferentemente hidrocloruro de cisteína. La principal desventaja de los sistemas de matriz es que los mismos exhiben generalmente un perfil de liberación de primer orden. Es decir, inicialmente las partículas de fármaco localizadas en la superficie del comprimido se disolverán y el fármaco se liberará rápidamente. Después de ello, las partículas de fármaco a distancias sucesivamente crecientes de la superficie del comprimido se disolverán y se liberarán por difusión en los poros al exterior del comprimido. Así, la distancia de difusión del fármaco aumentará a medida que transcurre el proceso de liberación. Normalmente se prefiere que se obtenga un perfil de liberación de orden cero o de orden próximo a cero en lugar de un perfil de liberación de primer orden. El sistema de liberación de orden cero proporciona una tasa de liberación constante del fármaco a lo largo de un periodo definido de tiempo. El mismo se utiliza fundamentalmente para fármacos con semividas cortas a fin de que los niveles constantes en sangre de los compuestos fármaco activos puedan mantenerse con menores dosis.
La patente U.S. nº 6.589.553 y la publicación internacional nº WO 02/062299 describen supuestamente una formulación de cápsulas para administración una vez al día con dos poblaciones de pelets recubiertos, cada uno de los cuales libera hidrocloruro de bupropión a un pH diferente. Una población de pelets está recubierta para liberar el fármaco a un pH correspondiente a aproximadamente 4,8 e inferior. La liberación del fármaco por esta población de pelets se espera que ocurra en el tracto gastrointestinal superior. La otra población de pelets está recubierta para liberar el fármaco a un pH de 7 y superior. La liberación de bupropión a partir de esta población se espera que ocurra en el tracto gastrointestinal inferior. En un ejemplo presentado, la biodisponibilidad relativa de bupropión a Zyban® era solamente 40% en términos de relación Cmax y solamente 80% en términos de relación AUC0-inf. En otro ejemplo representado, la biodisponibilidad relativa de bupropión a Zyban® era solamente 48% y el 59% en términos de Cmax y AUC0-inf. Las referencias describen adicionalmente la introducción de una tercera población de pelets activos sin recubrimiento, que supuestamente dan como resultado una modificación adicional y mejora de la liberación de bupropión. Basándose en el perfil plasmático medio concentración-tiempo representado en las Figuras 3 y 4 de estas referencias, no es fácilmente evidente que la introducción de los pelets activos sin recubrimiento pudiera dar como resultado una formulación bioequivalente de una sola vez al día (el producto de referencia es Zyban®). Asimismo, ninguna de las dos referencias presenta dato alguno de estabilidad del fármaco.
La patente US nº 6.033.686 describe un comprimido de liberación controlada, exento de estabilizador y exento de agente formador de poros que comprende un núcleo constituido esencialmente por hidrocloruro de bupropión, un aglomerante y un lubricante; y un recubrimiento que comprende un polímero insoluble...
Reivindicaciones:
1. Comprimido de liberación modificada que comprende:
en el que el comprimido de liberación modificada es bioequivalente y exhibe un perfil de disolución tal que después de aproximadamente 2 horas se libera no más de aproximadamente 20% del contenido de bupropión, después de aproximadamente 4 horas se libera 15% a 45% del contenido de bupropión, después de aproximadamente 8 horas se libera 40% a 90% del contenido de bupropión y después de aproximadamente 16 horas se libera no menos de aproximadamente 80% del contenido de bupropión.
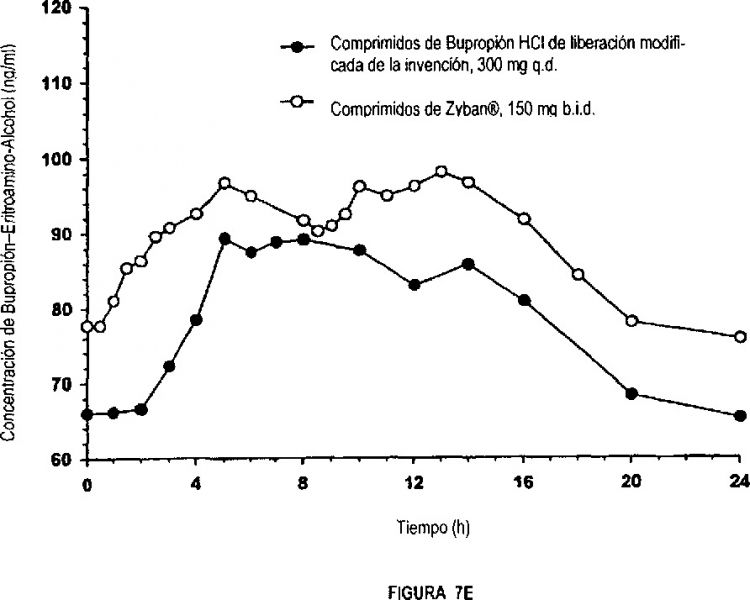
2. Comprimido de liberación modificada según la reivindicación 1, en el que dicho comprimido de liberación modificada es bioequivalente y en el que dicho comprimido de liberación modificada, cuando se administra a un paciente que precisa dicha administración en estado de ayuno proporciona una Cmax de bupropión en el plasma sanguíneo a entre 3 horas y 8 horas (Tmax) después de la administración del comprimido de liberación modificada, preferentemente a aproximadamente 5 horas (Tmax) después de la administración del comprimido de liberación modificada.
3. Comprimido de liberación modificada según la reivindicación 1 ó 2, en el que dicha barrera de humedad no funciona como un recubrimiento entérico como se define por un test USP que requiere, para un comprimido con recubrimiento de capa entérica, cuando se introduce en HCl 0,1 N durante 1 hora, que la cantidad total del fármaco liberada del núcleo no exceda de 10% y se libere no menos de 75% del fármaco a los 45 minutos en tampón de pH 6,8.
4. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 3, en el que la aplicación de la barrera de humedad al comprimido con recubrimiento de liberación controlada da como resultado un aumento de peso total no superior a aproximadamente 6% con relación al peso seco del comprimido, preferentemente no superior a aproximadamente 2,5% con relación al peso seco del comprimido.
5. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 4, en el que el polímero entérico es un polímero acrílico.
6. Comprimido de liberación modificada según la reivindicación 5, en el que dicho polímero acrílico es un copolímero de ácido metacrílico tipo C.
7. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 6, seleccionada de entre un comprimido que comprende aproximadamente 150 mg de dicha sal farmacéuticamente aceptable de bupropión, y la cantidad de dicho polímero entérico está comprendida entre 1% y 3% del peso seco del comprimido y comprende 55% a 70% del peso seco de la barrera de humedad, o un comprimido que comprende aproximadamente 300 mg de dicha sal farmacéuticamente aceptable de bupropión, y la cantidad de dicho polímero entérico está comprendida entre 1,5% y 3,0% del peso seco del comprimido y comprende desde 30% a 90% del peso seco de la barrera de humedad.
8. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 7 en el que el polímero es Eudragit® L 30D-55.
9. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 8 en el que dicho comprimido exhibe un perfil de disolución seleccionado de entre:
10. Comprimido de liberación modificada según cualquiera de las reivindicaciones anteriores, en el que dicha sal farmacéuticamente aceptable de bupropión es el hidrocloruro de bupropión.
11. Comprimido de liberación modificada según cualquiera de las reivindicaciones anteriores, en el que dicho bupropión está presente en por lo menos aproximadamente 94% en peso del núcleo.
12. Comprimido de liberación modificada según cualquiera de las reivindicaciones anteriores, en el que dichos excipientes convencionales comprenden además un aglomerante y un lubricante.
13. Comprimido de liberación modificada según la reivindicación 12, en el que dicho aglomerante está presente desde 1% a 6% en peso del peso seco del núcleo, preferentemente aproximadamente 3% en peso del peso seco del núcleo.
14. Comprimido de liberación modificada según las reivindicaciones 12 ó 13, en el que dicho aglomerante se selecciona de entre el grupo constituido por almidón modificado, gelatina, polivinilpirrolidona, derivados de celulosa, poli(alcohol vinílico) y cualquier combinación de los mismos.
15. Comprimido de liberación modificada según cualquiera de las reivindicaciones 12 a 14, en el que dicho lubricante está presente desde 1% a 6% en peso del peso seco del núcleo, preferentemente aproximadamente 3% en peso del peso seco del núcleo.
16. Comprimido de liberación modificada según cualquiera de las reivindicaciones 12 a 15, en el que dicho lubricante se selecciona de entre el grupo constituido por behenato de glicerilo, ácido esteárico, aceites vegetales hidrogenados y cualquier combinación de los mismos.
17. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 16, en el que dicho polímero formador de película permeable al agua insoluble en agua está presente en 35% a 60% en peso de dicho peso seco de recubrimiento de liberación controlada, preferentemente aproximadamente 50% en peso de dicho peso seco del recubrimiento de liberación controlada, más preferentemente aproximadamente 45% en peso del peso seco del comprimido.
18. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 17, en el que dicho polímero formador de película permeable al agua insoluble en agua se selecciona de entre el grupo constituido por un éter de celulosa, un éster de celulosa, poli(alcohol vinílico) y cualquier combinación de los mismos.
19. Comprimido de liberación modificada según la reivindicación 18, en el que dicho éter de celulosa se selecciona de entre el grupo constituido por etilcelulosa grado PR100, etilcelulosa grado PR20 y cualquier combinación de las mismas.
20. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 19, en el que dicho plastificante está presente desde 6% a 30% en peso de dicho peso seco del recubrimiento de liberación controlada, preferentemente aproximadamente 12% en peso de dicho peso seco del recubrimiento de liberación controlada.
21. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 20, en el que dicho plastificante se selecciona de entre el grupo constituido por polioles, ésteres orgánicos, aceites/glicéridos y cualquier combinación de los mismos.
22. Comprimido de liberación modificada según la reivindicación 21, en el que dicho poliol es el polietilenglicol 1450.
23. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 22, en el que dicho polímero soluble en agua está presente desde 25% a 50% en peso de dicho peso seco del recubrimiento de liberación controlada, preferentemente aproximadamente 43% en peso de dicho peso seco del recubrimiento de liberación controlada.
24. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 23, en el que dicho polímero soluble en agua se selecciona de entre el grupo constituido por polivinilpirrolidona, hidroxipropil-metilcelulosa, hidroxipropil-celulosa, y cualquier combinación de las mismas.
25. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 24, en el que la relación del polímero formador de película permeable al agua insoluble en agua:plastificante:polímero soluble en agua se selecciona de entre las relaciones de:
26. Comprimido de liberación modificada según cualquiera de las reivindicaciones anteriores, en el que el peso aumentado después de la aplicación del recubrimiento de liberación controlada se selecciona de entre el grupo de
27. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 26, en el que dicho polímero entérico está presente desde 30% a 90% en peso del peso seco de la barrera de humedad, preferentemente aproximadamente 66% en peso del peso seco de la barrera de humedad.
28. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 27, en el que dicho plastificante está presente en desde 1% a 30% en peso del peso seco de la barrera de humedad, preferentemente aproximadamente 10% en peso del peso seco de la barrera de humedad.
29. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 28, en el que dicho plastificante es una combinación de un éster orgánico y un poliol.
30. Comprimido de liberación modificada según la reivindicación 29, en el que dicha combinación de plastificante se encuentra en una proporción de 1 parte de éster orgánico a 2 partes de poliol.
31. Comprimido de liberación modificada según la reivindicación 29 ó 30, en el que dicho éster orgánico es el citrato de trietilo y dicho poliol es el polietilenglicol 1450.
32. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 31, en el que dicho mejorador de la permeación está presente en aproximadamente 25% en peso del peso seco de la barrera de humedad.
33. Comprimido de liberación modificada según las reivindicaciones 1 a 32, en el que dicho mejorador de la permeación se selecciona de entre el grupo constituido por dióxido de silicio, silicio coloidal, lactosa, polímeros hidrófilos, cloruro de sodio, óxido de aluminio, óxido de aluminio coloidal, sílice, celulosa microcristalina y cualquier combinación de los mismos.
34. Comprimido de liberación modificada según cualquiera de las reivindicaciones 1 a 33, en el que dicho polímero entérico, plastificante y mejorador de la permeación están presentes en una relación de aproximadamente 13:2:5.
35. Comprimido de liberación modificada según cualquiera de las reivindicaciones 10 a 34, que comprende:
en el que dicho comprimido de liberación modificada es bioequivalente y en el que dicho comprimido de liberación modificada, cuando se administra a un paciente que precisa dicha administración en estado de ayuno proporciona una Cmax de bupropión en el plasma sanguíneo a entre 3 horas y 8 horas (Tmax) después de la administración del comprimido de liberación modificada, preferentemente a aproximadamente 5 horas (Tmax) después de la administración del comprimido de liberación modificada.
36. Comprimido de liberación modificada según la reivindicación 35, en el que dicho comprimido de liberación modificada es bioequivalente y en el que dicho comprimido de liberación modificada, cuando se administra a un paciente que precisa dicha administración en estado de ayuno proporciona una Cmax de bupropión comprendido entre 60 ng/ml y 280 ng/ml en el plasma sanguíneo a aproximadamente 5 horas (Tmax) después de la administración de una dosis de 300 mg una sola vez al día de dicho comprimido de hidrocloruro de bupropión de liberación modificada o una dosis de una vez al día de 2 x 150 mg de dicho comprimido de hidrocloruro de bupropión de liberación modificada.
37. Comprimido de liberación modificada según la reivindicación 36, en el que dicho comprimido de liberación modificada es bioequivalente y se selecciona de entre un comprimido de liberación modificada que, cuando se administra a un paciente que se encuentra en necesidad de dicha administración en estado de ayuno exhibe una AUC(0-t) para bupropión de 800 ng.h/ml a 2.850 ng.h/ml después de la administración de una sola dosis al día de 300 mg de dicho comprimido de hidrocloruro de bupropión de liberación modificada o una dosis una vez al día de 2 x 150 mg de dicho comprimido de hidrocloruro de bupropión de liberación modificada, o un comprimido de liberación modificada cuando se administra a un paciente que se encuentra en necesidad de dicha administración en estado de ayuno exhibe una AUC(0-inf) para bupropión de 840 ng.h/ml a 3.000 ng.h/ml después de administración de una dosis de 300 mg una sola vez al día de dicho comprimido de hidrocloruro de bupropión de liberación modificada o una dosis de una vez al día de 2 x 150 mg de dicho comprimido de hidrocloruro de bupropión de liberación modificada.
38. Comprimido de liberación modificada según la reivindicación 36 ó 37, en el que dicho comprimido de liberación modificada administrado como una dosis diaria de 2 x 150 mg o una dosis administrada una vez al día de 300 mg a un paciente que se encuentra en necesidad de dicha administración no exhibe un efecto de los alimentos.
Patentes similares o relacionadas:
FORMULACIÓN DE RESAGILINA DE LIBERACIÓN RETARDADA, del 26 de Diciembre de 2011, de TEVA PHARMACEUTICAL INDUSTRIES LTD.: Citrato de rasagilina
FORMULACIÓN DE RABEPRAZOL, del 21 de Diciembre de 2011, de LEK PHARMACEUTICALS D.D.: Composición farmacéutica que comprende un núcleo de comprimido que comprende rabeprazol sódico, hidróxido de calcio, al menos un excipiente farmacéuticamente […]
 FORMULACIÓN EN COMPRIMIDOS DE LIBERACIÓN EXTENDIDA QUE CONTIENE PRAMIPEXOL O UNA DE SUS SALES FARMACÉUTICAMENTE ACEPTABLES, MÉTODO PARA SU FABRICACIÓN Y SU USO, del 30 de Marzo de 2011, de BOEHRINGER INGELHEIM INTERNATIONAL GMBH: Una formulación en comprimidos de liberación extendida que comprende pramipexol, o su sal farmacéuticamente aceptable, en una matriz que comprende al menos […]
FORMULACIÓN EN COMPRIMIDOS DE LIBERACIÓN EXTENDIDA QUE CONTIENE PRAMIPEXOL O UNA DE SUS SALES FARMACÉUTICAMENTE ACEPTABLES, MÉTODO PARA SU FABRICACIÓN Y SU USO, del 30 de Marzo de 2011, de BOEHRINGER INGELHEIM INTERNATIONAL GMBH: Una formulación en comprimidos de liberación extendida que comprende pramipexol, o su sal farmacéuticamente aceptable, en una matriz que comprende al menos […]
FORMULACIONES QUE CONTIENEN SALICILATOS Y SU USO PARA TRATAR LA ENFERMEDAD INFLAMATORIA INTESTINAL, del 4 de Marzo de 2011, de AGI THERAPEUTICS RESEARCH LIMITED: Una composición farmacéutica de liberación modificada que comprende: un salicilato y/o ácido salicílico elegidos entre ácido 4-aminosalicílico, ácido 5- profármacos de […]
FORMULACIONES FARMACEUTICAS DE COMPUESTOS INHIBIDORES DE AMILOIDE, del 18 de Noviembre de 2009, de BELLUS HEALTH (INTERNATIONAL) LIMITED: Formulación que comprende una sustancia que se selecciona a partir de ácido 3-amino-1-propanosulfónico y sus sales farmacéuticamente aceptables, […]
 METODO PARA EL RECUBRIMIENTO DE FARMACOS Y COMPOSICIONES OBTENIDAS A PARTIR DEL MISMO, del 1 de Noviembre de 2008, de CENAVISA S.L.: Método para el recubrimiento de fármacos y composiciones obtenidas a partir del mismo. La presente invención está relacionada con el campo de la tecnologia farmacéutica, […]
METODO PARA EL RECUBRIMIENTO DE FARMACOS Y COMPOSICIONES OBTENIDAS A PARTIR DEL MISMO, del 1 de Noviembre de 2008, de CENAVISA S.L.: Método para el recubrimiento de fármacos y composiciones obtenidas a partir del mismo. La presente invención está relacionada con el campo de la tecnologia farmacéutica, […]
Formulación de vitamina D de liberación modificada estabilizada y método de administración de la misma, del 22 de Julio de 2020, de EirGen Pharma Ltd: Una formulacion oral de liberacion controlada de un compuesto de vitamina D que comprende uno o ambos de 25- hidroxivitamina D2 y 25-hidroxivitamina D3, la formulacion […]
Composición farmacéutica que comprende un agente antipsicótico atípico y método para su preparación, del 15 de Julio de 2020, de PHARMATHEN S.A.: Comprimido de liberación controlada de Paliperidona en forma de comprimido de varias capas que comprende: a) un núcleo de matriz que comprende […]