CEBADORES, SONDAS, PROCEDIMIENTOS Y USOS DE LOS MISMOS PARA LA DETECCIÓN DE MYCOBACTERIIUM KANSASII.
Un oligonucleótido específico de Mycobacterium kansasii para la detección de Mycobacterium kansasii que comprende una secuencia de nucleótidos representada en la SEQ ID Núm.
1 o una secuencia de nucleótidos complementaria a la secuencia de nucleótidos
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/JP2006/309514.
Solicitante: WAKO PURE CHEMICAL INDUSTRIES, LTD.
Nacionalidad solicitante: Japón.
Dirección: 1-2, DOSHOMACHI 3-CHOME, CHUO-KU OSAKA-SHI, OSAKA 540-8605 JAPON.
Inventor/es: ISHIKAWA, TOMOKAZU.
Fecha de Publicación: .
Fecha Solicitud PCT: 11 de Mayo de 2006.
Clasificación Internacional de Patentes:
- C12Q1/04 QUIMICA; METALURGIA. › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12Q PROCESOS DE MEDIDA, INVESTIGACION O ANALISIS EN LOS QUE INTERVIENEN ENZIMAS, ÁCIDOS NUCLEICOS O MICROORGANISMOS (ensayos inmunológicos G01N 33/53 ); COMPOSICIONES O PAPELES REACTIVOS PARA ESTE FIN; PROCESOS PARA PREPARAR ESTAS COMPOSICIONES; PROCESOS DE CONTROL SENSIBLES A LAS CONDICIONES DEL MEDIO EN LOS PROCESOS MICROBIOLOGICOS O ENZIMOLOGICOS. › C12Q 1/00 Procesos de medida, investigación o análisis en los que intervienen enzimas, ácidos nucleicos o microorganismos (aparatos de medida, investigación o análisis con medios de medida o detección de las condiciones del medio, p. ej. contadores de colonias, C12M 1/34 ); Composiciones para este fin; Procesos para preparar estas composiciones. › Determinación de la presencia o del tipo de microorganismo; Empleo de medios selectivos para la investigación o análisis de antibióticos o bactericidas; Composiciones para este fin que contienen un indicador químico.
- C12Q1/68M10B
- G01N33/569D4
Clasificación PCT:
- C12N15/09 C12 […] › C12N MICROORGANISMOS O ENZIMAS; COMPOSICIONES QUE LOS CONTIENEN; PROPAGACION, CULTIVO O CONSERVACION DE MICROORGANISMOS; TECNICAS DE MUTACION O DE INGENIERIA GENETICA; MEDIOS DE CULTIVO (medios para ensayos microbiológicos C12Q 1/00). › C12N 15/00 Técnicas de mutación o de ingeniería genética; ADN o ARN relacionado con la ingeniería genética, vectores, p. ej. plásmidos, o su aislamiento, su preparación o su purificación; Utilización de huéspedes para ello (mutantes o microorganismos modificados por ingeniería genética C12N 1/00, C12N 5/00, C12N 7/00; nuevas plantas en sí A01H; reproducción de plantas por técnicas de cultivo de tejidos A01H 4/00; nuevas razas animales en sí A01K 67/00; utilización de preparaciones medicinales que contienen material genético que es introducido en células del cuerpo humano para tratar enfermedades genéticas, terapia génica A61K 48/00; péptidos en general C07K). › Tecnología del ADN recombinante.
- C12Q1/04 C12Q 1/00 […] › Determinación de la presencia o del tipo de microorganismo; Empleo de medios selectivos para la investigación o análisis de antibióticos o bactericidas; Composiciones para este fin que contienen un indicador químico.
- C12Q1/68 C12Q 1/00 […] › en los que intervienen ácidos nucleicos.
Países PCT: Austria, Bélgica, Suiza, Alemania, Dinamarca, España, Francia, Reino Unido, Grecia, Italia, Liechtensein, Luxemburgo, Países Bajos, Suecia, Mónaco, Portugal, Irlanda, Eslovenia, Finlandia, Rumania, Chipre, Lituania, Letonia.
PDF original: ES-2357902_T3.pdf
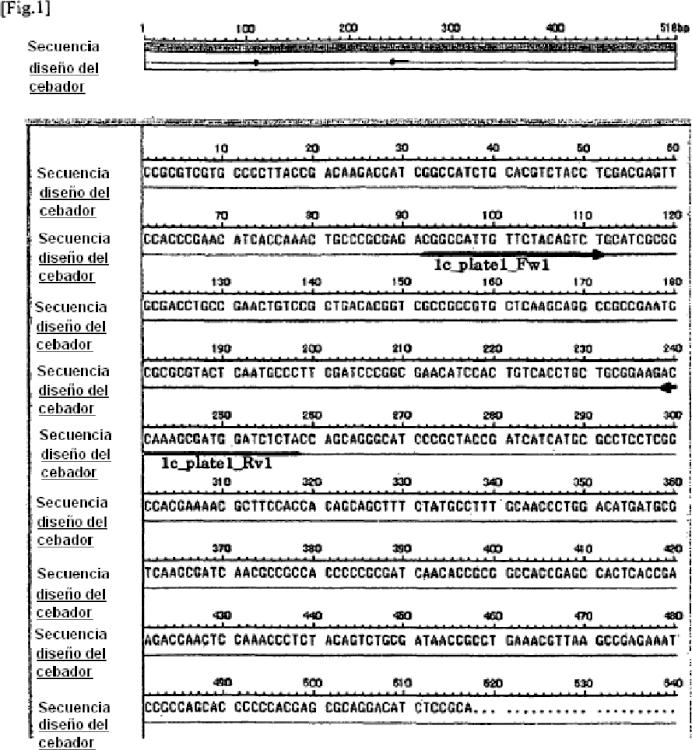
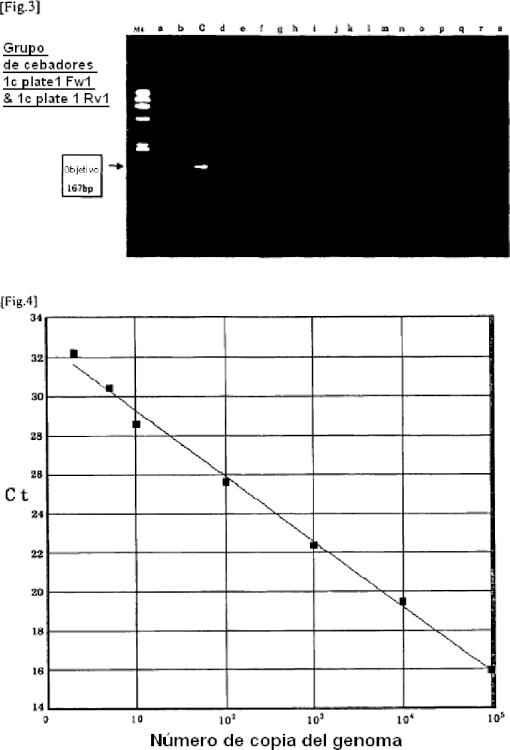
Fragmento de la descripción:
CAMPO TÉCNICO
La presente invención se refiere a un procedimiento para detectar y/o identificar M. kansasii (Mycobacterium kansasii, en adelante descrita M. kansasii) a través del uso de amplificación de ácido nucleico y un sistema de detección del mismo en un ensayo clínico de laboratorio.
ANTECEDENTES DE LA INVENCIÓN
Mycobacterium no tuberculosa (NTM) es un bacilo gram positivo que tiene características de ácido-alcohol resistentes clasificado en el género Mycobacterium, y es un tipo de bacteria ácido-alcohol resistente distinta al complejo de tuberculosis y Mycobacterium leprae.
Entre la mycobacterium no tuberculosa, es sabido que la cepa bacteriana clínicamente problemática incluye Mycobacterium kansasii, Mycobacterium marinum, Mycobacterium gordonae, Mycobacterium szulgai, Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium xenopi, Mycobacterium fortuitum, Mycobacterium chelonei, Mycobacterium abscessus, y etc. En particular, las enfermedades infecciosas afectadas por 2 tipos de bacterias, M. kansasii y complejo M. avium, representan el 90% o más de la totalidad de las enfermedades por mycobacterium no tuberculoso.
En general, se sostiene que la mycobacterium no tuberculosa es inofensiva para un individuo saludable, sin embargo, en ocasiones poco frecuentes, puede infectar a seres humanos y causar enfermedades por mycobacterium no tuberculosa. Particularmente, en los individuos inmunocomprometidos tales como pacientes infectados con el virus del SIDA puede ser un agente causativo de infecciones serias. En el pasado, las enfermedades por mycobacterium no tuberculosa han sido un trastorno poco frecuente, sin embargo, en los últimos años, la incidencia de infección demuestra una tendencia en alza, y, por lo tanto, se ha deseado ampliamente el desarrollo de un procedimiento para discriminar bacterias tuberculosas de mycobacterium no tuberculosa en un período de tiempo breve. Además, dado que el procedimiento para detectar/diagnosticar M. avium y M. intracellulare por amplificación de ácido nucleico ha sido aprobado para su inclusión en la cobertura del seguro médico y después se ha propagado rápidamente por el país, su significancia diagnóstica es, por supuesto, muy grande.
Dado que la mayoría de las mycobacteria no tuberculosas tienen una resistencia a agentes antituberculosos, cuando se sospecha que un paciente tiene una infección por una bacteria ácido-alcohol resistente, la diferencia en el diagnóstico referente a si la enfermedad es una enfermedad por mycobacterium tuberculosa o no tuberculosa será muy importante a la hora de decidir el curso del tratamiento. Además, ya que el procedimiento para el tratamiento de las enfermedades causadas por mycobacteria no tuberculosas puede variar para cada tipo de bacteria, la identificación de las especies bacterianas también será muy importante. Sin embargo, dado que la enfermedad por mycobacterium no tuberculosa no tiene un síntoma clínico específico, es muy difícil diferenciar la enfermedad por mycobacterium tuberculosa de la enfermedad por mycobacterium no tuberculosa por observación clínica y manifestación histopatológica, más aún lo es especificar las especies de la mycobacterium no tuberculosa. Por lo tanto, el diagnóstico referente a si la enfermedad es una enfermedad por mycobacterium tuberculosa o no tuberculosa debe llevarse a cabo por la identificación de la bacteria infectada.
En un diagnóstico típico, en primer lugar, se examina el frotis del esputo. Mediante este ensayo, puede reconocerse únicamente “la bacteria ácido-alcohol resistente positiva", y no puede lograrse la diferenciación de la bacteria tuberculosa de la mycobacterium no tuberculosa. Por lo tanto, cuando el examen del frotis del esputo es positivo, se lleva cabo un examen de un cultivo bacteriano por cultivo en aislamiento sobre un medio de cultivo especificado tal como medio de Ogawa para diferenciar la bacteria tuberculosa de la mycobacterium no tuberculosa. Además, a través de exámenes químicos adicionales, se identifica la especie de la bacteria. Sin embargo, en general, el crecimiento de la bacteria perteneciente al género Mycobacterium es lento, y su cultivo lleva un tiempo considerable. Por consiguiente, en los procedimientos básicos del procedimiento convencional que incluye examen del esputo y examen del cultivo, el resultado del diagnóstico por parte del cultivo en aislamiento que informa si la bacteria es tuberculosa o no, demora de 3 a 4 semanas. Además, existe otro problema, dado que requiere de 2 a 3 semanas adicionales para completar varios ensayos bioquímicos para la identificación de especies bacterianas.
Además, la identificación de M. kansasii también se lleva a cabo por ensayos bioquímicos. En el procedimiento principal para identificar M. kansasii por ensayos bioquímicos, se utiliza la propiedad específica para producir pigmentos cuando la bacteria se expone a la luz. Sin embargo, dado que otras especies pertenecientes al género Mycobacterium muestran las mismas propiedades que M. kansasii, la identificación de M. kansasii por su propiedad de coloración es en general problemática.
En los años recientes, se han desarrollado tecnologías para detectar bacterias a un nivel genético. Por ejemplo, se ha estudiado como un medio útil una técnica de diagnóstico que utiliza tecnología de amplificación de ácido nucleico tal como reacción en cadena de la polimerasa (PCR). Este procedimiento tiene ventajas de alta sensibilidad; varias células de las bacterias son suficientes para la detección; la detección puede completarse en un período de tiempo breve (de 4 días como máximo). Sin embargo, en el procedimiento de PCR usual, se detectan por igual tanto células vivas como células muertas. Además, dado que el procedimiento se juzga positivo independientemente del tamaño del recuento de bacterias, y que el número de las bacterias es desconocido, el diagnóstico de si la infección es positiva o no se proporcionará con incertidumbre. Además, el procedimiento tiene un problema, dado que cuenta con una sensibilidad demasiado alta, existe la posibilidad de falsos juicios positivos.
Respecto a M. kansasii, existe un estudio que informa que se obtuvo una sonda de ADN (pMKI-9) a partir de una librería genómica de M. kansasii (Documento de No Patente 1). Esta sonda de ADN (pMK1-9) puede formar un híbrido complementario con el ADN de M. kansasii, pero esta sonda también puede formar un híbrido con otras especies de mycobacteria, y no es específica a M. kansasii.
Asimismo, existe un estudio focalizado en el uso de una sonda de ADN comercialmente disponible (ACCUPROBE™, GenProbe, San Diego, CA) que puede hibridizarse específicamente con la sonda pMK1-9 y el gen de ARNr de M. kansasii para la identificación de M. kansasii (Documento de No Patente 2). Sin embargo, en este estudio, se ha informado que tanto la sonda pMK1-9 como la sonda de ADN comercialmente disponible (ACCU-PROBE™) fueron incapaces de detectar un número considerable de tipos de cepa de M. kartsasii.
Además, existe otro estudio en el que se evaluó la sonda de ADN comercialmente disponible (ACCUPROBE™) para la detección de M. kansasii (Documento de No Patente 3). Los investigadores de este estudio informaron que si bien ACCU-PROBE™ es 100% especie específico, y no muestra ninguna reacción cruzada con otras especies de M. kansasii, sólo podría detectarse en este experimento un 73% de las especies de M. kansasii.
Existe un informe que describe que una sonda de formación de ADN híbrido (p6123) específica a M. kansasii se ha purificado con un aislado clínico de M. kansasii (Documento de No Patente 4). La sonda (p6123) fue capaz de hibridizarse con todas las cepas de M. kansasii utilizadas en este experimento que incluyen un subgrupo que no reaccionó con una sonda de ADN (pMK1-9) informado por Ross et al. La Patente de los Estados Unidos Núm.
5.500.341 (Documento de Patente 2) ha desvelado un cebador de amplificación específico de M. kansasii purificado con una sonda p6123.
Además, B. Boddinghaus et al. han desvelado un oligonucleótido específico de Mycobacterium purificado con ARNr 16S, que específica se prolifera e hibridiza con ADN de mycobacterium (Documento de No Patente 5).
Además, por ejemplo, también se ha estudiado la identificación de la región del ADN efectiva para detectar M. kansasii (por ejemplo, Documento de Patente 1), sin embargo, la presente situación indica no se ha establecido el procedimiento de diagnóstico específico de M. kansasii.... [Seguir leyendo]
Reivindicaciones:
1. Un oligonucleótido específico de Mycobacterium kansasii para la detección de Mycobacterium kansasii que comprende una secuencia de nucleótidos representada en la SEQ ID Núm. 1 o una secuencia de nucleótidos complementaria a la secuencia de nucleótidos.
2. Un cebador para detectar Mycobacterium kansasii, que comprende un oligonucleótido que comprende una secuencia de nucleótidos seleccionada de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, en la que el oligonucleótido es una parte de un oligonucleótido que comprende una secuencia de nucleótidos representada en la SEQ ID Núm. 1, o
un oligonucleótido que comprende una secuencia de nucleótidos complementaria a la secuencia de nucleótidos seleccionada a partir de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, en la que el oligonucleótido es una parte de un oligonucleótido que comprende una secuencia de nucleótidos complementaria a la secuencia de nucleótidos representada en la SEQ ID Núm. 1, y en la que el oligonucleótido es capaz de hibridizarse con una secuencia de nucleótidos del gen de Mycobacterium kansasii.
3. El cebador de acuerdo con la reivindicación 2, en el que el cebador está marcado con una secuencia marcadora.
4. El cebador de acuerdo con la reivindicación 3, en el que la sustancia marcadora se selecciona de un radioisótopo, una enzima, una sustancia fluorescente o biotina.
5. Una sonda para detectar Mycobacterium kansasii, que comprende un oligonucleótido que comprende una secuencia de nucleótidos seleccionada de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, 53 a 56 y 80, en la que el oligonucleótido es una parte de un oligonucleótido que comprende una secuencia de nucleótidos representada en la SEQ ID Núm.1, o
un oligonucleótido que comprende una secuencia de nucleótidos complementaria a la secuencia de nucleótidos seleccionada a partir de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, 53 a 56 y 80, en la que el oligonucleótido es una parte de un oligonucleótido que comprende una secuencia de nucleótidos complementaria a la secuencia de nucleótidos representada en la SEQ ID Núm. 1, y en el que el oligonucleótido es capaz de hibridizarse con una secuencia de nucleótidos del gen de Mycobacterium kansasii.
6. La sonda de acuerdo con la reivindicación 5, en la que la sonda está marcada con una sustancia marcadora.
7. La sonda de acuerdo con la reivindicación 6, en la que la sustancia marcadora se selecciona de un radioisótopo, una enzima, una sustancia fluorescente o biotina.
8. La sonda de acuerdo con la reivindicación 5, en la que el extremo terminal 5' está marcado con un tinte fluorescente reportero y el extremo terminal 3' está marcado con un tinte desactivante.
9. Un procedimiento para detectar Mycobacterium kansasii que comprende utilizar un oligonucleótido que comprende una secuencia de nucleótidos seleccionada de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, en la que el oligonucleótido es una parte de un oligonucleótido que comprende una secuencia de nucleótidos representada en la SEQ ID Núm. 1, o
un oligonucleótido que comprende una secuencia de nucleótidos complementaria a la secuencia de nucleótidos seleccionada de la secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, en la que el oligonucleótido es una parte de un oligonucleótido que comprende una secuencia de nucleótidos complementaria a la secuencia de nucleótidos representada en la SEQ ID Núm.1, y
en la que el oligonucleótido es capaz de hibridizarse con una secuencia de nucleótidos de Mycobacterium kansasii, como un cebador, y/o un oligonucleótido que comprende una secuencia de nucleótidos seleccionada de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, 53 a 56 y 80, en la que el oligonucleótido es una parte de un oligonucleótido que comprende una secuencia de nucleótidos representada en la SEQ ID Núm.1, o
un oligonucleótido que comprende una secuencia de nucleótidos complementaria a la secuencia de nucleótidos seleccionada de la secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, 53 a 56 y 80, en el que el oligonucleótido es una parte de un oligonucleótido que comprende una secuencia de nucleótidos complementaria a la secuencia de nucleótidos representada en la SEQ ID Núm.1, y
en la que el oligonucleótido es capaz de hibridizarse con una secuencia de nucleótidos de Mycobacterium kansasii, como una sonda.
10. El procedimiento de detección de acuerdo con la reivindicación 9, que comprende;
llevar a cabo una reacción de amplificación de ácido nucleico con el uso como un cebador de un oligonucleótido que comprende una secuencia de nucleótidos seleccionada de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, o una secuencia de nucleótidos complementaria a la secuencia de nucleótidos seleccionada de la secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, en la que el oligonucleótido es capaz de hibridizarse con una secuencia de nucleótidos del gen de Mycobacterium kansasii; y
utilizar un ácido nucleico en una muestra como una plantilla; y
detectar un producto de extensión del cebador.
11. El procedimiento de detección de acuerdo con la reivindicación 9, en el que el procedimiento comprende los siguientes procedimientos:
(1) llevar a cabo una reacción de amplificación de ácido nucleico con el uso como un cebador de un oligonucleótido que comprende una secuencia de nucleótidos seleccionada de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, o una secuencia de nucleótidos complementaria a la secuencia de nucleótidos seleccionada de la secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, en la que el oligonucleótido es capaz de hibridizarse con una secuencia de nucleótidos del gen de Mycobacterium kansasii, y utilizar un ácido nucleico en una muestra como una plantilla; y
(2) llevar a cabo electroforesis del producto de extensión del cebador obtenido en el apartado anterior (1), y detectar Mycobacterium kansasii sobre la base del resultado obtenido.
12. El procedimiento de detección de acuerdo con la reivindicación 11, que además comprende después de llevar a cabo electroforesis, confirmar una fracción del producto de extensión del cebador que tenga el número objetivo de pares de base en la fracción electroforética obtenida, en la que el número objetivo de pares de base sea el número de pares de base del producto de extensión del cebador, que se espera sea replicado por PCR por el uso del cebador directo y el cebador inverso.
13. El procedimiento de detección de acuerdo con la reivindicación 11, que además comprende, después de la electroforesis, hibridizar la fracción electroforética obtenida con una sonda marcada preparada por el marcado de un oligonucleótido con una sustancia marcadora, en la que el oligonucleótido comprende una secuencia de nucleótidos seleccionada de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, 53 a 56 y 80, o una secuencia de nucleótidos complementaria a la secuencia de nucleótidos seleccionada de la secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, 53 a 56 y 80, y es capaz de hibridizarse con la secuencia de nucleótidos del gen de Mycobacterium kansasii;
detectar una señal derivada de la sonda marcada, y
determinar la muestra que está confirmada que tiene una fracción hibridizada con la sonda marcada mencionada con anterioridad, que es positiva.
14. El procedimiento de detección de acuerdo con la reivindicación 13, en el que la sustancia marcadora se selecciona de un radioisótopo, una enzima, una sustancia fluorescente, una sustancia luminiscente y biotina.
15. El procedimiento para detectar Mycobacterium kansasii de acuerdo con la reivindicación 9, en el que el cebador está marcado con una secuencia marcadora; y
el procedimiento incluye un procedimiento para llevar a cabo una reacción en cadena de polimerasa por el uso de un cebador marcado y un ácido nucleico en una muestra como una plantilla, y medir una señal derivada del producto de extensión del cebador obtenido.
16. El procedimiento de detección de acuerdo con la reivindicación 15, en el que la sustancia marcadora se selecciona de un radioisótopo, una enzima, una sustancia fluorescente, una sustancia luminiscente y biotina.
17. El procedimiento de detección de acuerdo con la reivindicación 15, que además comprende después de llevar a cabo la reacción en cadena de polimerasa, eliminar un cebador marcado libre, y medir la señal derivada del producto de extensión del cebador.
18. El procedimiento de detección de acuerdo con la reivindicación 17, en el que el cebador marcado libre se elimina por la eliminación de un sobrenadante después de la precipitación del producto de extensión del cebador en el producto de reacción que se obtiene llevando a cabo una reacción en cadena de la polimerasa.
19. El procedimiento de detección de acuerdo con la reivindicación 17, en el que el cebador marcado libre se elimina por el tratamiento del producto de reacción, que se obtiene llevando a cabo una reacción en cadena de la polimerasa, con cromatografía en gel.
20. El procedimiento para detectar Mycobacterium kansasii de acuerdo con la reivindicación 9, que comprende;
hibridizar una sonda marcada preparada por la marcación del oligonucleótido que comprende una secuencia de nucleótidos seleccionada de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, 53 a 56 y 80, o una secuencia de nucleótidos complementaria a la secuencia de nucleótidos seleccionada de la secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, 53 a 56 y 80, en la que el oligonucleótido es capaz de hibridizarse con la secuencia de nucleótidos del gen de Mycobacterium kansasii con una sustancia marcadora,
eliminar una sonda marcada libre, y
detectar una señal derivada de un complejo hibridizado.
21. El procedimiento de detección de acuerdo con la reivindicación 20, en el que la sustancia marcadora se selecciona de un radioisótopo, una enzima, una sustancia fluorescente, una sustancia luminiscente y biotina.
22. Un kit para detectar Mycobacterium kansasii, que comprende un oligonucleótido que comprende una secuencia de nucleótidos seleccionada de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, o una secuencia de nucleótidos complementaria a la secuencia de nucleótidos seleccionada de la secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12 como un cebador, y/o un oligonucleótido que comprende una secuencia de nucleótidos seleccionada de una secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, 53 a 56 y 80, o una secuencia de nucleótidos complementaria a la secuencia de nucleótidos seleccionada de la secuencia de nucleótidos representada en la SEQ ID Núm. 5 a 12, 53 a 56 y 80 como una sonda, en la que el oligonucleótido es capaz de hibridizarse con la secuencia de nucleótidos del gen de Mycobacterium kansasii gene, como un cebador y/o una sonda.
23. El kit de acuerdo con la reivindicación 22, en el que el cebador y/o la sonda están marcados con una sustancia marcadora.
24. El kit de acuerdo con la reivindicación 23, en el que la sustancia marcadora se selecciona de un radioisótopo, una enzima, una sustancia fluorescente, una sustancia luminiscente y biotina.
25. El kit de acuerdo con la reivindicación 23, en el que la sonda es aquella en la que el extremo terminal 5' está marcado con un tinte fluorescente reportero y el extremo terminal 3' está marcado con un tinte inactivador.
26. Uso de un oligonucleótido para diseñar un cebador o una sonda para la detección de Mycobacterium kansasii, en el que el oligonucleótido comprende una secuencia de nucleótidos representada en la SEQ ID Núm. 1, o una secuencia complementaria a la secuencia de nucleótidos, y que es capaz de hibridizarse con la secuencia de nucleótidos del gen de Mycobacterium kansasii.
Patentes similares o relacionadas:
Método, sistema y producto del programa informático para determinar la presencia de microorganismos e identificar dichos microorganismos, del 29 de Julio de 2020, de BIOMERIEUX: Un método para determinar la presencia de al menos un microorganismo determinado en una placa de Petri que comprende una o más colonias de microorganismos y un medio de […]
Preparación de células biológicas sobre soportes de muestras para espectrometría de masas para una ionización por desorción, del 13 de Mayo de 2020, de Bruker Daltonik GmbH: Procedimiento para la preparación de proteínas a partir de muestras de células biológicas no purificadas sobre un soporte de muestras para una determinación mediante […]
Disposición de detector para botellas de cultivo de sangre con sensores colorimétricos, del 13 de Mayo de 2020, de BIOMERIEUX, INC.: Un dispositivo de detección que comprende: una botella de cultivo de sangre que incorpora un sensor colorimétrico sujeto a cambio de color debido a un cambio […]
Método mejorado para la determinación de microorganismos, del 6 de Mayo de 2020, de STORA ENSO OYJ: Un método para determinar el contenido de microorganismos en un material que comprende celulosa en la industria de la pulpa y el papel que comprende las etapas de: […]
Procedimiento para detectar bacterias coliformes contenidas en la leche, del 15 de Abril de 2020, de ASAHI KASEI KABUSHIKI KAISHA: Procedimiento para lisar bacterias coliformes contenidas en la leche, que comprende la etapa de mezclar un agente de lisis que contiene lisozima, un surfactante aniónico […]
Protocolos de rastreo de lotes múltiples de composiciones modulares para la detección de patógenos, contaminantes microbianos y/o constituyentes, del 25 de Marzo de 2020, de INSTITUTE FOR ENVIRONMENTAL HEALTH, INC: Un procedimiento de muestreo, ensayo y validación de lotes de ensayo, que comprende: a) recoger una pluralidad de porciones de cada una de una pluralidad de lotes de ensayo, […]
Composiciones y procedimientos para identificar especies de Ehrlichia, del 18 de Marzo de 2020, de Abaxis, Inc: Un procedimiento de identificación de las especies de Ehrlichia que infectan a un sujeto, si están presentes, comprendiendo el procedimiento: poner en contacto una […]
Métodos y grupos, del 11 de Marzo de 2020, de Genome Research Limited: Un método para la identificación de cepas aisladas bacterianas adecuadas para su uso en bacterioterapia, comprendiendo el método: (i) preparar una suspensión […]