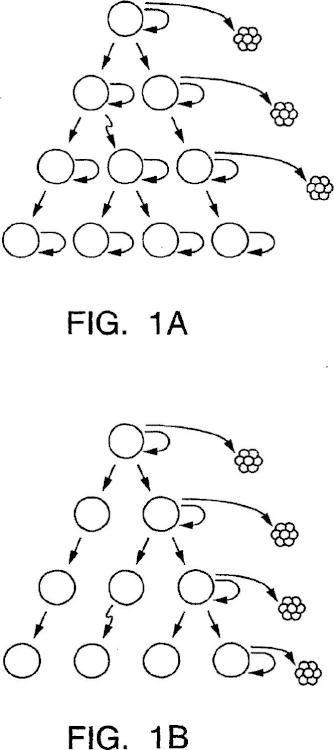
Aislamiento y uso de células madre de tumores sólidos.
Un método in vitro para enriquecer una población de células madre de tumor sólido,
que comprende las etapas de:
(a) disociar un tumor sólido de cáncer epitelial;
(b) poner en contacto las células disociadas con un primer reactivo que se une a CD44 y un segundo reactivo que se une a CD24; y
(c) seleccionar células que se unen al primer reactivo y que no se unen de manera detectable o que se unen débilmente al segundo reactivo, en donde las células seleccionadas están enriquecidas en células madre tumorales.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/US2001/024243.
Solicitante: THE REGENTS OF THE UNIVERSITY OF MICHIGAN.
Nacionalidad solicitante: Estados Unidos de América.
Dirección: 1600 Huron Parkway, 2nd Floor Ann Arbor, MI 48109-2590 ESTADOS UNIDOS DE AMERICA.
Inventor/es: CLARKE, MICHAEL, F., AL-HAJJ,Muhammad, MORRISON,SEAN J, WICHA,MAX S.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- A01K67/027 NECESIDADES CORRIENTES DE LA VIDA. › A01 AGRICULTURA; SILVICULTURA; CRIA; CAZA; CAPTURA; PESCA. › A01K CRÍA DE ANIMALES; AVICULTURA; APICULTURA; PISCICULTURA; PESCA; ANIMALES PARA CRIA O REPRODUCCIÓN, NO PREVISTOS EN OTRO LUGAR; NUEVAS VARIEDADES DE ANIMALES. › A01K 67/00 Cría u obtención de animales, no prevista en otro lugar; Nuevas razas de animales. › Nuevas razas de vertebrados.
- A61K39/00 A […] › A61 CIENCIAS MEDICAS O VETERINARIAS; HIGIENE. › A61K PREPARACIONES DE USO MEDICO, DENTAL O PARA EL ASEO (dispositivos o métodos especialmente concebidos para conferir a los productos farmacéuticos una forma física o de administración particular A61J 3/00; aspectos químicos o utilización de substancias químicas para, la desodorización del aire, la desinfección o la esterilización, vendas, apósitos, almohadillas absorbentes o de los artículos para su realización A61L; composiciones a base de jabón C11D). › Preparaciones medicinales que contienen antígenos o anticuerpos (materiales para ensayos inmunológicos G01N 33/53).
- A61K39/395 A61K […] › A61K 39/00 Preparaciones medicinales que contienen antígenos o anticuerpos (materiales para ensayos inmunológicos G01N 33/53). › Anticuerpos (aglutininas A61K 38/36 ); Inmunoglobulinas; Inmunosuero, p. ej. suero antilinfocitario.
- C07K14/705 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07K PEPTIDOS (péptidos que contienen β -anillos lactamas C07D; ipéptidos cíclicos que no tienen en su molécula ningún otro enlace peptídico más que los que forman su ciclo, p. ej. piperazina diones-2,5, C07D; alcaloides del cornezuelo del centeno de tipo péptido cíclico C07D 519/02; proteínas monocelulares, enzimas C12N; procedimientos de obtención de péptidos por ingeniería genética C12N 15/00). › C07K 14/00 Péptidos con más de 20 aminoácidos; Gastrinas; Somatostatinas; Melanotropinas; Sus derivados. › Receptores; Antígenos celulares de superficie; Determinantes celulares de superficie.
- C07K14/71 C07K 14/00 […] › para factores de crecimiento; para reguladores de crecimiento.
- C07K16/28 C07K […] › C07K 16/00 Inmunoglobulinas, p. ej. anticuerpos mono o policlonales. › contra receptores, antígenos celulares de superficie o determinantes celulares de superficie.
- C07K16/30 C07K 16/00 […] › de células tumorales.
- C12N5/095 C […] › C12 BIOQUIMICA; CERVEZA; BEBIDAS ALCOHOLICAS; VINO; VINAGRE; MICROBIOLOGIA; ENZIMOLOGIA; TECNICAS DE MUTACION O DE GENETICA. › C12N MICROORGANISMOS O ENZIMAS; COMPOSICIONES QUE LOS CONTIENEN; PROPAGACION, CULTIVO O CONSERVACION DE MICROORGANISMOS; TECNICAS DE MUTACION O DE INGENIERIA GENETICA; MEDIOS DE CULTIVO (medios para ensayos microbiológicos C12Q 1/00). › C12N 5/00 Células no diferenciadas humanas, animales o vegetales, p. ej. líneas celulares; Tejidos; Su cultivo o conservación; Medios de cultivo para este fin (reproducción de plantas por técnicas de cultivo de tejidos A01H 4/00). › Células madre; Células progenitoras.
- C12Q1/68 C12 […] › C12Q PROCESOS DE MEDIDA, INVESTIGACION O ANALISIS EN LOS QUE INTERVIENEN ENZIMAS, ÁCIDOS NUCLEICOS O MICROORGANISMOS (ensayos inmunológicos G01N 33/53 ); COMPOSICIONES O PAPELES REACTIVOS PARA ESTE FIN; PROCESOS PARA PREPARAR ESTAS COMPOSICIONES; PROCESOS DE CONTROL SENSIBLES A LAS CONDICIONES DEL MEDIO EN LOS PROCESOS MICROBIOLOGICOS O ENZIMOLOGICOS. › C12Q 1/00 Procesos de medida, investigación o análisis en los que intervienen enzimas, ácidos nucleicos o microorganismos (aparatos de medida, investigación o análisis con medios de medida o detección de las condiciones del medio, p. ej. contadores de colonias, C12M 1/34 ); Composiciones para este fin; Procesos para preparar estas composiciones. › en los que intervienen ácidos nucleicos.
- G01N33/50 FISICA. › G01 METROLOGIA; ENSAYOS. › G01N INVESTIGACION O ANALISIS DE MATERIALES POR DETERMINACION DE SUS PROPIEDADES QUIMICAS O FISICAS (procedimientos de medida, de investigación o de análisis diferentes de los ensayos inmunológicos, en los que intervienen enzimas o microorganismos C12M, C12Q). › G01N 33/00 Investigación o análisis de materiales por métodos específicos no cubiertos por los grupos G01N 1/00 - G01N 31/00. › Análisis químico de material biológico, p. ej. de sangre o de orina; Ensayos mediante métodos en los que interviene la formación de uniones bioespecíficas con grupos coordinadores; Ensayos inmunológicos (procedimientos de medida o ensayos diferentes de los procedimientos inmunológicos en los que intervienen enzimas o microorganismos, composiciones o papeles reactivos a este efecto, procedimientos para preparar estas composiciones, procedimientos de control sensibles a las condiciones del medio en los procedimientos microbiológicos o enzimáticos C12Q).
- G01N33/574 G01N 33/00 […] › para el cáncer.
PDF original: ES-2553796_T3.pdf

Patentes similares o relacionadas:
Eliminación de impurezas de cultivos celulares residuales, del 29 de Julio de 2020, de NOVARTIS AG: Un método para eliminar la Proteína Nuclear (NP) de la Gripe de una preparación que comprende proteínas del virus de la gripe de interés que incluyen hemaglutinina […]
 Cepas de Bordetella vivas atenuadas como vacuna de dosis única contra la tos ferina, del 29 de Julio de 2020, de INSTITUT PASTEUR DE LILLE: Una vacuna que comprende una cepa viva, atenuada y mutada de Bordetella pertussis que comprende al menos una mutación del gen (ptx) de la toxina […]
Cepas de Bordetella vivas atenuadas como vacuna de dosis única contra la tos ferina, del 29 de Julio de 2020, de INSTITUT PASTEUR DE LILLE: Una vacuna que comprende una cepa viva, atenuada y mutada de Bordetella pertussis que comprende al menos una mutación del gen (ptx) de la toxina […]
Método para producir inmunoconjugados de anticuerpo-SN-38 con un enlazador CL2A, del 22 de Julio de 2020, de IMMUNOMEDICS, INC.: Un método para producir un compuesto, CL2A-SN-38, que presenta la estructura, **(Ver fórmula)** que comprende realizar un esquema de reacción como el que se muestra: **(Ver […]
Composición de vacuna que contiene un adyuvante sintético, del 22 de Julio de 2020, de INFECTIOUS DISEASE RESEARCH INSTITUTE: Una composición farmacéutica que comprende: un adyuvante lípido de glucopiranosilo (GLA), que tiene la fórmula: **(Ver fórmula)** en la que: […]
Arenavirus trisegmentados como vectores de vacunas, del 22 de Julio de 2020, de UNIVERSITE DE GENEVE: Una partícula de arenavirus trisegmentada infecciosa y competente para la replicación que comprende un segmento L y dos segmentos S, en donde uno de los dos segmentos […]
Inmunoterapia novedosa contra diversos tumores, entre ellos tumores cerebrales y neuronales, del 22 de Julio de 2020, de IMMATICS BIOTECHNOLOGIES GMBH: Péptido que comprende una secuencia de aminoácidos acorde con la SEQ ID N.º 19, en que dicho péptido tiene una longitud total de entre 9 y 16 aminoácidos.
Métodos mejorados para la preparación de escualeno, del 15 de Julio de 2020, de NOVARTIS AG: Un procedimiento para la preparación de escualeno a partir de una composición que comprende escualeno a partir de una fuente animal, dicho procedimiento comprendiendo […]
Formulaciones estables que contienen anticuerpos anti-PCSK9, del 15 de Julio de 2020, de AMGEN INC.: Una formulación estable que comprende un anticuerpo monoclonal que se une específicamente a PCSK9, en donde PCSK9 comprende los aminoácidos de la SEQ ID NO: […]