Un proceso para la producción de ácido metacrílico y sus derivados y los polímeros producidos a partir de los mismos.
Un proceso para la producción de ácido metacrílico mediante la descarboxilación catalizada por base de al menos un ácido dicarboxílico seleccionado a partir de ácido itacónico,
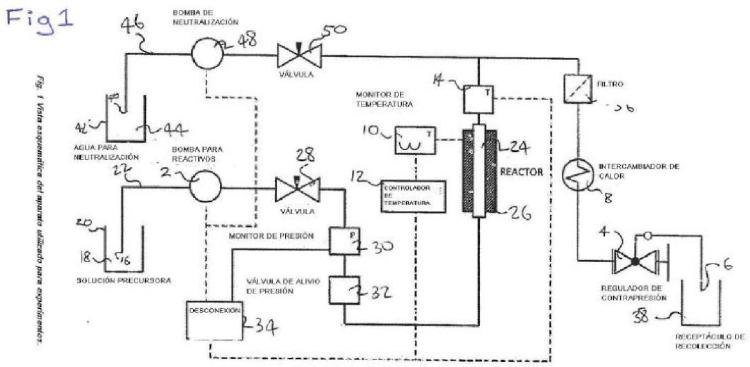
citracónico o mesacónico o mezclas de los mismos, en donde la descarboxilación se lleva a cabo a más de 240 y hasta 275ºC.
Tipo: Patente Internacional (Tratado de Cooperación de Patentes). Resumen de patente/invención. Número de Solicitud: PCT/GB2011/052271.
Solicitante: LUCITE INTERNATIONAL UK LIMITED.
Nacionalidad solicitante: Reino Unido.
Dirección: Cumberland House, 15-17 Cumberland Place Southampton, Hampshire SO15 2BG REINO UNIDO.
Inventor/es: JOHNSON, DAVID WILLIAM, EASTHAM, GRAHAM RONALD, POLIAKOFF, MARTYN, HUDDLE,THOMAS ANDREW.
Fecha de Publicación: .
Clasificación Internacional de Patentes:
- C07C51/38 QUIMICA; METALURGIA. › C07 QUIMICA ORGANICA. › C07C COMPUESTOS ACICLICOS O CARBOCICLICOS (compuestos macromoleculares C08; producción de compuestos orgánicos por electrolisiso electroforesis C25B 3/00, C25B 7/00). › C07C 51/00 Preparación de ácidos carboxílicos o sus sales, haluros o anhídridos. › por descarboxilación.
- C07C57/04 C07C […] › C07C 57/00 Compuestos insaturados que tienen grupos carboxilo unidos a átomos de carbono acíclicos. › Acido acrílico; Acido metacrílico.
- C07C67/03 C07C […] › C07C 67/00 Preparación de ésteres de ácidos carboxílicos. › por reacción de un grupo éster con un grupo hidroxilo.
- C08F20/06 C […] › C08 COMPUESTOS MACROMOLECULARES ORGANICOS; SU PREPARACION O PRODUCCION QUIMICA; COMPOSICIONES BASADAS EN COMPUESTOS MACROMOLECULARES. › C08F COMPUESTOS MACROMOLECULARES OBTENIDOS POR REACCIONES QUE IMPLICAN UNICAMENTE ENLACES INSATURADOS CARBONO - CARBONO (producción de mezclas de hidrocarburos líquidos a partir de hidrocarburos de número reducido de átomos de carbono, p. ej. por oligomerización, C10G 50/00; Procesos de fermentación o procesos que utilizan enzimas para la síntesis de un compuesto químico dado o de una composición dada, o para la separación de isómeros ópticos a partir de una mezcla racémica C12P; polimerización por injerto de monómeros, que contienen uniones insaturadas carbono-carbono, sobre fibras, hilos, hilados, tejidos o artículos fibrosos hechos de estas materias D06M 14/00). › C08F 20/00 Homopolímeros o copolímeros de compuestos que tienen uno o más radicales alifáticos insaturados, teniendo cada uno solamente un enlace doble carbono-carbono, y estando solamente uno terminado por un radical carboxilo o una sal, anhídrido, éster, amida, imida o nitrilo del mismo. › Acido acrílico; Acido metacrílico; Sus sales metálicas o de amonio.
- C08F20/10 C08F 20/00 […] › Esteres.
- C08F20/14 C08F 20/00 […] › Esteres de metilo.
- C08L33/06 C08 […] › C08L COMPOSICIONES DE COMPUESTOS MACROMOLECULARES (composiciones basadas en monómeros polimerizables C08F, C08G; pinturas, tintas, barnices, colorantes, pulimentos, adhesivos D01F; filamentos o fibras artificiales D06). › C08L 33/00 Composiciones de homopolímeros o copolímeros de compuestos que tienen uno o más radicales alifáticos insaturados, teniendo solamente cada uno un enlace doble carbono-carbono, y estando solamente uno terminado por un solo radical carboxilo, o sus sales, anhídridos, ésteres, amidas, imidas o nitrilos; Composiciones de los derivados de tales polímeros. › de ésteres que contienen solamente carbono, hidrógeno y oxígeno, estando los átomos de oxígeno solamente presentes como parte de un radical carboxilo.
PDF original: ES-2552981_T3.pdf
Patentes similares o relacionadas:
Proceso para la producción de biodiésel y productos relacionados, del 15 de Julio de 2020, de ARGENT ENERGY (UK) LIMITED: Un proceso para producir biodiésel a partir de una mezcla, comprendiendo dicho proceso las etapas de: (i) proporcionar la mezcla a un recipiente de reacción […]
Proceso mejorado de preparación de ésteres alquílicos de ácido graso (biodiésel) a partir de aceites de triglicéridos usando catalizadores básicos sólidos ecológicos, del 13 de Mayo de 2020, de Council of Scientific & Industrial Research An Indian registered body incorporated under the Registration of Societies Act (Act XXI of 1860): Un procedimiento de preparación de ésteres alquílicos de ácidos grasos (FAAE) a partir de aceites de triglicéridos seleccionados del grupo […]
Ésteres epoxidados de color reducido a partir de grasas y aceites naturales epoxidados, del 11 de Marzo de 2020, de ARCHER-DANIELS-MIDLAND COMPANY: Un proceso para preparar a) un éster de ácido graso epoxidado de color reducido a partir de una grasa o aceite natural epoxidado o b) un éster de ácido graso […]
Proceso para despolimerizar cutina, del 11 de Marzo de 2020, de Apeel Technology Inc: Un método para preparar monómeros, oligómeros, ésteres de los mismos o combinaciones de los mismos derivados de la cutina a partir de material vegetal que contiene […]
Proceso y aparato para purificar una mezcla de residuos grasos y productos relacionados incluyendo combustibles, del 26 de Febrero de 2020, de ARGENT ENERGY (UK) LIMITED: Un proceso para purificar una mezcla que comprende grasa, aceite y/o grasa de cocina, comprendiendo dicho proceso las etapas de: (i) calentamiento inicial de la mezcla […]
Composición de biodiésel y proceso y productos relacionados, del 19 de Febrero de 2020, de ARGENT ENERGY (UK) LIMITED: Una composición de biodiésel que comprende una mezcla de ésteres, en donde la mezcla de ésteres comprende de 7% en peso a 10,5% en peso de octadecanoato de metilo, […]
Proceso para la producción de biodiésel y productos relacionados, del 23 de Octubre de 2019, de ARGENT ENERGY (UK) LIMITED: Un proceso para esterificar una mezcla que comprende entre el 10% y 20% en peso de ácidos grasos libres (FFAs), comprendiendo dicho proceso las etapas de: […]
Procedimiento de preparación de éster alquílico de ácido graso utilizando grasa, del 22 de Octubre de 2019, de SK CHEMICALS CO., LTD.: Procedimiento de preparación de éster alquílico de ácido graso para combustibles biodiesel, que comprende las etapas de: preparar ácido graso y glicerina mediante una […]