17 patentes, modelos y diseños de RANBAXY LABORATORIES, LTD.
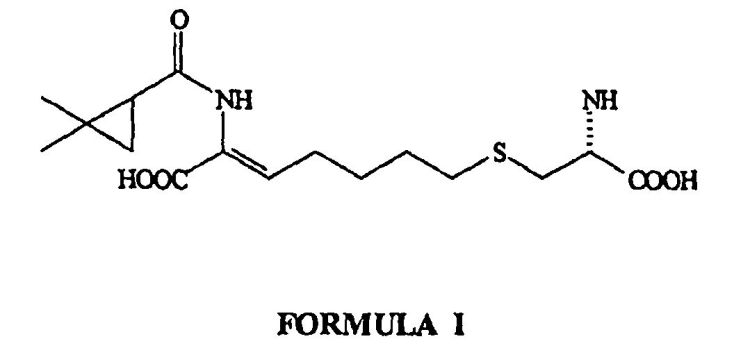
PROCEDIMIENTO PARA LA PREPARACION DE CILASTATINA.
Sección de la CIP Química y metalurgia
(16/10/2007). Ver ilustración. Inventor/es: KUMAR, YATENDRA, TYAGI, OM, DUTT, SRIVASTAVA,TUSHAR,KUMAR,C/O DR. NARENDRA KUMAR, PANDEY,ANAND. Clasificación: C07C323/59, C07C319/28.
Proceso para la purificación de la cilastatina, que comprende poner en contacto una solución de cilastatina cruda con una resina adsorbente no iónica y la recuperación de cilastatina pura a partir de una solución de la misma.
DERIVADOS AZABICICLO UTILIZADOS COMO ANTAGONISTAS DE LOS RECEPTORES MUSCARINICOS.
(16/05/2007) Compuestos que tienen la estructura de la fórmula I: y sus sales farmacéuticamente aceptables, solvatos farmacéuticamente aceptables, estéres, enantiómeros, diastereómeros, N-óxidos o polímorfos, en los que Ar representa un anillo de arilo o heteroarilo y tiene 1-2 hetero átomos, los anillos de arilo o de heteroarilo pueden ser no substituidos o substituidos por uno a tres substituyentes seleccionados independientemente entre alquilo inferior (C1-C4), perhalo alquilo inferior (C1-C4), ciano, hidroxi, nitro, alcoxi inferior (C1-C4), perhalo alcoxi inferior (C1-C4), amino no substituido, N-alquilo inferior (C1-C4) o -arilo amino, amino carbonilo o N-alquilo inferior (C1-C4) o -arilo amino carbonilo; R1 representa hidrógeno, hidroxi, hidroxi metilo, amino substituido o no substituido, alcoxi, carbamoilo o halógeno; R2 representa alquilo, un anillo…
PROCEDIMIENTO PARA CONTROLAR LA MEZCLA DE HIDRATOS DE LA SAL DISODICA DE FOSFLUCONAZOL.
(01/03/2007) Un procedimiento para la preparación de una mezcla de hidratos estable de la sal disódica de fosfluconazol, o una composición que comprende dicho compuesto, que comprende: a) proporcionar una cantidad de una mezcla acuosa que contiene la sal disódica de fosfluconazol o una composición del mismo en un recipiente adecuado en un equipo de secado por congelación; b) reducir la temperatura en el equipo para llevarlo aproximadamente a la congelación y solidificación eutéctica; c) reducir la presión en el equipo hasta por debajo de la presión de vapor de saturación (SVP) de agua sobre hielo a la temperatura del hielo; d) mantener el equipo a una presión por debajo de la SVP y, de forma opcional, aumentar la temperatura en el equipo para facilitar la sublimación, hasta que todo el hielo haya sublimado; e) mantener el equipo en las condiciones de presión…
COMPOSICION FARMACEUTICA ORAL DE CEFPODOXIMA PROXETILO.
Sección de la CIP Necesidades corrientes de la vida
(16/03/2006). Inventor/es: MATHUR, RAJEEV, SHANKAR, MALIK, RAJIV, MALHOTRA, MUKTA. Clasificación: A61K9/00, A61K9/20, A61K31/545.
Una composición farmacéutica para su administración oral que comprende una cantidad farmacéuticamente eficaz de cefpodoxima proxetilo adsorbida a un transportador farmacéutico en la que la cefpodoxima proxetilo presenta un tamaño medio de partícula inferior a los 30 micrómetros y una distribución de tamaño de partícula tal que al menos un 90% de las partículas son inferiores a 75 micrómetros y el área superficial es superior a los 1, 3 m2/g.
PROCESO DE PREPARACION DE REPAGLINIDA.
(01/11/2005) La presente invención proporciona el proceso de la preparación de la repaglinida de Fórmula I que comprende: a) la reacción de la (S)-amina de Fórmula II con un ácido carboxílico protegido de Fórmula IV donde R es un grupo protector, con la presencia de cloruro de pivaloil y una base, y b) la eliminación del grupo protector para obtener repaglinida. El grupo protector R en el compuesto de Fórmula IV es cualquier grupo protector de ácido carboxílico que se elimina fácilmente, como el metil, el etil, el tertbutil, el benzil, el p-nitrobenzil, el p-metoxibenzil, y sus similares. La reacción se lleva a cabo en presencia de una base adecuada que puede ser orgánica o inorgánica. Ejemplos de bases orgánicas…
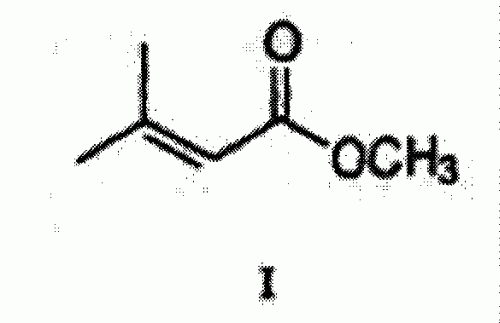
PROCESO DE PREPARACION DE LA ISOTRETINOINA.
Sección de la CIP Química y metalurgia
(16/10/2005). Ver ilustración. Inventor/es: SALMAN, MOHAMMAD, KUMAR, NARESH, KAUL, VIJAY, KUMAR, BABU, J., SURESH. Clasificación: C07C403/20.
Un proceso para la preparación de la isotretinoína en una reacción de una única etapa que comprende la condensación del dienolato de metil-3, 3-dimetil acrilato de Fórmula I: con el acetaldehído de ß-ionilideno de Fórmula II: en un disolvente adecuado a una temperatura entre -60 ºC y -80 ºC durante 1 2 horas para producir la lactona intermedia de Fórmula III: y aumentando la temperatura de la mezcla de la reacción hasta los 25 ºC a 45 ºC durante 1 24 horas, y a continuación con un tratamiento acídico acuoso para formar la isotretinoína de Fórmula IV:.
DOIFICACION ORAL BIO-APROVECHABLE DE LORATADINE.
Sección de la CIP Necesidades corrientes de la vida
(01/05/2005). Inventor/es: MALIK, RAJIV, KUMAR, PANANCHUKUNATH MANOJ, GUPTA, DINSHEET. Clasificación: A61K31/4545, A61K9/14.
Forma de dosificación oral bio-aprovechable de loratadine que comprende loratadine que presenta un tamaño medio de partículas comprendido entre aprox. 0, 1 micrones a aprox. 15 micrones y que presenta un área superficial comprendida entre 1 y 2, 5 m2/g.
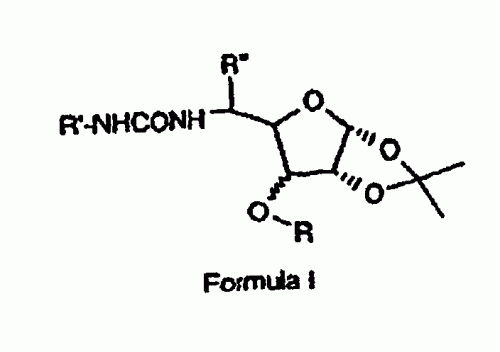
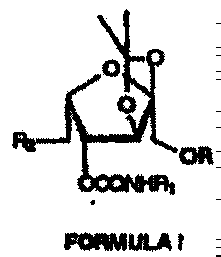
DERIVADOS DE MONOSACARIDOS COMO INHIBIDORES DE LA ADHESION CELULAR.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(01/03/2005). Ver ilustración. Inventor/es: ARORA, SUDERSHAN K., TANWAR, MADAN P., GUPTA, JANG BAHADUR, SHARMA, GEETA. Clasificación: A61K31/70, C07H9/04.
Compuesto que tiene la estructura de Fórmula I **(Fórmula)** y sus sales, ésteres, enantiómeros, diastereómeros, N-óxidos, amidas farmacéuticamente aceptables, en la que R es un grupo alquilo, alqueno, alquino (cadena lineal o ramificada), arilo, arilo sustituido o alquilarilo de C1 a C15, R es un grupo SO2C6H5, SO2C6H4CH3-p, o SO2C6H4Cl-p, fenilo o fenilo sustituido, representado como C6H4-R-p, siendo R Cl, NO2, OCH3, CH3, CH2COOH, CH2COOCH3, CH2COLDVP, CH2CODVP, CH2COVP, en el que LDVP, DVP y VP representan un tetrapéptido (Leucil-aspartil-valil-prolil) , un tripéptido (aspartil-valil-prolil) y un dipéptido (valilprolil), respectivamente, R es H o CH3.
PROCESO DE PREPPARACION DE UNA FORMA DE CEFUROXIMO AXETIL AMORFA, PURA Y DE CALIDAD FARMACOPEA.
Sección de la CIP Química y metalurgia
(01/03/2005). Ver ilustración. Inventor/es: HANDA, VIJAY, KUMAR, TYAGI, OM, DUTT, RAY, UTTAM, KUMAR. Clasificación: C07D501/00.
Proceso de preparación de una forma de cefuroximo axetil amorfa, pura y de calidad farmacopea que comprende disolver cefuroximo axetil cristalino en ácido acético incluyendo, como mínimo, el 5% v/v de agua y recuperar dicha forma amorfa de cefuroximo axetil de la disolución mediante la adición de agua, siendo vertida la disolución en el agua y estando el agua presente en un volumen suficiente para precipitar dicho cefuroximo axetil amorfo.
PROCEDIMIENTO EFICAZ EN COSTE PARA LA METILACION SELECTIVA DE DERIVADOS DE ERITROMICINA A.
Sección de la CIP Química y metalurgia
(01/11/2004). Inventor/es: SALMAN, MOHAMMAD, KUMAR, NARESH, RAY, PURNA CHANDRA, GANGAKHEDKAR, KIRAN KUMAR, LAL DORWAL, HARISH NIRANJAN. Clasificación: C07H17/08.
Procedimiento para la metilación selectiva de un grupo hidroxi en la posición 6 de un derivado de la eritromicina A que comprende la metilación del derivado de la eritromicina A con un agente metilador en una mezcla de tolueno y un disolvente aprótico polar en presencia de una base.
DERIVADOS DE 2,3-O-ISOPROPILIDENO DE MONOSACARIDOS Y SU UTILIZACION COMO INHIBIDORES DE LA ADHESION CELULAR.
Secciones de la CIP Necesidades corrientes de la vida Química y metalurgia
(16/05/2004). Ver ilustración. Inventor/es: ARORA, SUDERSHAN K., GUPTA, JANG BAHADUR, KISHORE, NAWAL, JOSHI, VISHWAS D. Clasificación: A61K31/70, C07H9/04.
Compuestos con la estructura de la Fórmula I: y sus sales, ésteres, enantiómeros, diastereómeros, N- óxidos y amidas farmacéuticamente aceptables, en la que R es alquilo, alqueno, alquino C1 a C15 (de cadena lineal o ramificada), aril, aril sustituido o alquilaril, y R1 es fenil o-, m- ó p-clorofenil, tolil, metoxifenil o nitrofenil, y R2 es H, pirrolidinil, piperidinil, morfolinil o hexametilenimino, o un radical de fórmula NHR3, en la que R3 es alquil, alquenil o alquinil C1 a C15 (de cadena lineal o ramificada), o un radical de Fórmula III: en la que n es un número entero, como máximo 5, y es un anillo heterocíclico de cinco, seis o siete elementos que contiene uno o más heteroátomos.
FORMULACION DE MATRIZ DE LIBERACION MODIFICADA DE CEFACLOR Y CEFALEXINA.
(16/04/2004) COMPOSICION FARMACEUTICA EN FORMA DE TABLETA PARA LIBERACION RETARDADA DE UN INGREDIENTE ACTIVO, QUE COMPRENDE CEFACLOR, CEFALEXINA SUS HIDRATOS, SALES O ESTERES FARMACEUTICAMENTE ACEPTABLES, COMO INGREDIENTE ACTIVO, Y UNA MEZCLA DE POLIMEROS HIDROFILOS DE DIFERENTES GRADOS DE VISCOSIDAD, SELECCIONADOS ENTRE EL GRUPO FORMADO POR AL MENOS UNA HIDROXIPROPIL-METILCELULOSA Y AL MENOS UNA HIDROXIPROPILCELULOSA. LA COMPOSICION CONTIENE TAMBIEN OPTATIVAMENTE UNO O MAS DILUYENTES SOLUBLES EN AGUA O DISPERSABLES EN AGUA. LAS CANTIDADES DE LOS POLIMEROS HIDROFILICOS Y DEL DILUYENTE SOLUBLE EN AGUA O DISPERSABLE EN AGUA PERMITE QUE EL INGREDIENTE…
SISTEMA DE SUMINISTRO CONTROLADO DE MEDICAMENTOS POR VIA ORAL CON CONTROL EN EL TIEMPO Y EN EL ESPACIO.
Sección de la CIP Necesidades corrientes de la vida
(16/04/2004). Inventor/es: SEN, HIMADRI, TALWAR, NARESH, STANIFORTH, JOHN, H. Clasificación: A61K9/52, A61K9/22.
Formulación para administración oral una vez al día en humanos para la liberación controlada de ciprofloxacina en el estómago o parte superior del intestino delgado, comprendiendo una cantidad farmacéuticamente efectiva de ciprofloxacina, alrededor de 0, 2% a 0, 5% de alginato sódico, alrededor de 0, 5 a 2, 0% de goma xantano, aproximadamente de 10, 0% a 25% de bicarbonato sódico, y aproximadamente de 5, 0% a 20% de polivinilpirrolidona reticulada, siendo dichos porcentajes peso/peso del compuesto, en el que la proporción en peso de alginato sódico con respecto a goma xantano está comprendida aproximadamente entre 1:1 y aproximadamente 1:10.
PROCEDIMIENTO PARA LA PREPARACION DE UNA FORMA DE DOSIFICACION ORAL BIODISPONIBLE DE AXETIL CEFUROXIMA.
Sección de la CIP Necesidades corrientes de la vida
(01/04/2004). Inventor/es: SEN, HIMADRI, SOMANI, JITENDRA KRISHAN, BHUSHAN, INDU. Clasificación: A61K9/20, A61K31/545.
Procedimiento para la preparación de una forma de dosificación oral que comprende la mezcla de axetil cefuroxima amorfa con axetil cefuroxima cristalina, tal que la axetil cefuroxima cristalina forma de 7 a 25 por ciento en peso de la cantidad total de axetil cefuroxima amorfa junto con axetil cefuroxima cristalina, de manera que la forma de dosificación comprendiendo la mezcla de axetil cefuroxima cristalina y amorfa mostró un perfil de biodisponibilidad comparable al de la axetil cefuroxima amorfa pura.
PRODUCTOS INTERMEDIARIOS CLAVE PARA LA PREPARACION DE SIMVASTATINA.
Sección de la CIP Química y metalurgia
(01/05/2002). Inventor/es: KUMAR, YATENDRA, KUMAR, S.M. DILEEP, KHANNA, JAG MOHAN, THAPER, RAJESH KUMAR, MISRA, SATYANANDA. Clasificación: C07D309/30, C07C235/30.
UN PROCESO PARA PREPARAR SIMVASTATINA A PARTIR DE LOVASTATINA O ACIDO MEVINOLINICO EN SU FORMA DE SAL INCLUYE TRATAR CUALQUIERA DE LOS MATERIALES DE PARTIDA CON CICLOPROPILO O BUTILAMIDA, ABRIENDOSE DE ESE MODO EL ANILLO PIRANONA CUANDO EL MATERIAL DE PARTIDA ES LOVASTATINA, AÑADIR UN GRUPO METILO A LA CADENA LATERAL 2 - METILBUTIRATO, Y POSTERIORMENTE CERRAR EL ANILLO PIRANONA ABIERTO PARA PRODUCIR SIMVASTATINA. EL PROCESO SE REALIZA SIN PROTEGER Y DESPROTEGER LOS DOS GRUPOS HIDROXI DEL ANILLO PIRANONA ABIERTO. EN UNA REALIZACION PREFERIDA, SE TRATA EL MATERIAL DE PARTIDA CON CICLOPROPILAMINA, LO CUAL PRODUCE SIMVASTATINA VIA EL NUEVO INTERMEDIO CICLOPROPILAMIDA DE LOVASTATINA.
PROCEDIMIENTO PARA LA FABRICACION DE SIMVASTATINA O ACIDO MEVINOLINICO.
Sección de la CIP Química y metalurgia
(16/12/2001). Inventor/es: KUMAR, YATENDRA, KUMAR, S.M. DILEEP, KHANNA, JAG MOHAN, THAPER, RAJESH KUMAR, MISRA, SATYANANDA. Clasificación: C07D309/30.
UN PROCESO PARA PREPARAR SIMVASTATINA A PARTIR DE LOVASTATINA O ACIDO MEVINOLINICO EN SU FORMA DE SAL INCLUYE TRATAR CUALQUIERA DE LOS MATERIALES DE PARTIDA CON CICLOPROPILO O BUTILAMIDA, ABRIENDOSE DE ESE MODO EL ANILLO PIRANONA CUANDO EL MATERIAL DE PARTIDA ES LOVASTATINA, AÑADIR UN GRUPO METILO A LA CADENA LATERAL 2 - METILBUTIRATO, Y POSTERIORMENTE CERRAR EL ANILLO PIRANONA ABIERTO PARA PRODUCIR SIMVASTATINA. EL PROCESO SE REALIZA SIN PROTEGER Y DESPROTEGER LOS DOS GRUPOS HIDROXI DEL ANILLO PIRANONA ABIERTO. EN UNA REALIZACION PREFERIDA, SE TRATA EL MATERIAL DE PARTIDA CON CICLOPROPILAMINA, LO CUAL PRODUCE SIMVASTATINA VIA EL NUEVO INTERMEDIO CICLOPROPILAMIDA DE LOVASTATINA.
PROCESO PARA LA PRODUCCION DE ALFA-6-DESOXITETRACICLINAS.
Sección de la CIP Química y metalurgia
(16/09/1993). Inventor/es: KHANNA, JAGMOHAN, BALA, KIRAN, GROVER, INDER PAL SINGH. Clasificación: C07C237/26.
UN PROCESO PARA LA HIDROGENACION DE UNA 6-METILENO-TETRACICLINA EN LA PRODUCCION DE ALFA-6-DESOXITETRACICLINA , PARTICULARMENTE EL ANTIBIOTICO DOXICICLINA, EN PRESENCIA DE UN COMPLEJO METALICO DE TRANSICION DE FORMULA MCLX(PPH3)Y, EN DONDE M ES PREFERIBLEMENTE CU,CO O NI, X ES 1 O 2, E Y ES DE 1 A 3, Y UNA TRAZA DE RODIO COMO METAL SOPORTADO, O BIEN, COMO SAL DE RODIO, COMO CATALIZADOR DE HIDROGENACION. EL DESEADO PRODUCTO ALFA-6-DESOXITETRACICLINA SE PRODUCE CON GRAN RENDIMIENTO Y ESTEREOESPECIFICIDAD. EL PROCESO REQUIERE EL USO DE COMPLEJOS METALICOS DE TRANSICION SUBSTANCIALMENTE BARATOS Y SOLO TRAZAS DE RODIO EN EL CATALIZADOR DE HIDROGENACION POR MOL DE LA 6-METILENO-TETRACICLINA HIDROGENADA.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}